Рефераты по авиации и космонавтике
Рефераты по административному праву
Рефераты по безопасности жизнедеятельности
Рефераты по арбитражному процессу
Рефераты по архитектуре
Рефераты по астрономии
Рефераты по банковскому делу
Рефераты по сексологии
Рефераты по информатике программированию
Рефераты по биологии
Рефераты по экономике
Рефераты по москвоведению
Рефераты по экологии
Краткое содержание произведений
Рефераты по физкультуре и спорту
Топики по английскому языку
Рефераты по математике
Рефераты по музыке
Остальные рефераты
Рефераты по биржевому делу
Рефераты по ботанике и сельскому хозяйству
Рефераты по бухгалтерскому учету и аудиту
Рефераты по валютным отношениям
Рефераты по ветеринарии
Рефераты для военной кафедры
Рефераты по географии
Рефераты по геодезии
Рефераты по геологии
Рефераты по геополитике
Рефераты по государству и праву
Рефераты по гражданскому праву и процессу
Рефераты по кредитованию
Рефераты по естествознанию
Рефераты по истории техники
Рефераты по журналистике
Рефераты по зоологии
Рефераты по инвестициям
Рефераты по информатике
Исторические личности
Рефераты по кибернетике
Рефераты по коммуникации и связи
Рефераты по косметологии
Рефераты по криминалистике
Рефераты по криминологии
Рефераты по науке и технике
Рефераты по кулинарии
Рефераты по культурологии
Рефераты по авиации и космонавтике
Рефераты по административному праву
Рефераты по безопасности жизнедеятельности
Рефераты по арбитражному процессу
Рефераты по архитектуре
Рефераты по астрономии
Рефераты по банковскому делу
Рефераты по сексологии
Рефераты по информатике программированию
Рефераты по биологии
Рефераты по экономике
Рефераты по москвоведению
Рефераты по экологии
Краткое содержание произведений
Рефераты по физкультуре и спорту
Топики по английскому языку
Рефераты по математике
Рефераты по музыке
Остальные рефераты
Рефераты по биржевому делу
Рефераты по ботанике и сельскому хозяйству
Рефераты по бухгалтерскому учету и аудиту
Рефераты по валютным отношениям
Рефераты по ветеринарии
Рефераты для военной кафедры
Рефераты по географии
Рефераты по геодезии
Рефераты по геологии
Рефераты по геополитике
Рефераты по государству и праву
Рефераты по гражданскому праву и процессу
Рефераты по кредитованию
Рефераты по естествознанию
Рефераты по истории техники
Рефераты по журналистике
Рефераты по зоологии
Рефераты по инвестициям
Рефераты по информатике
Исторические личности
Рефераты по кибернетике
Рефераты по коммуникации и связи
Рефераты по косметологии
Рефераты по криминалистике
Рефераты по криминологии
Рефераты по науке и технике
Рефераты по кулинарии
Рефераты по культурологии
Контрольная работа: Естествознание на молекулярном уровне
Контрольная работа: Естествознание на молекулярном уровне
ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ РФ
Российский Государственный торгово-экономический университет
Омский Институт
Контрольная работа
По курсу: «Концепции современного естествознания»
"Естествознание на молекулярном уровне"
Омск – 2008
Оглавление
Введение
1. История химии, алхимия
1.1 История алхимии
1.2 История химии
2. Неорганическая и органическая химия
2.1 Неорганическая химия
2.2 Органическая химия
3. Реакционная способность веществ, анализ и синтез
3.1 Реакционная способность веществ
3.2 Анализ
3.3 Органический синтез
4. Важнейшие химические связи и методы исследования веществ
5. Условия существования жизни, связи и функции живого вещества
6. Молекулярные основы эволюции (белки, нуклеиновые кислоты, первичный и вторичный генетический коды)
Заключение
Список используемой литературы
Введение
Развитие химических знаний стимулируется необходимостью получения человеком различных веществ для своей жизнедеятельности. Для этого приходилось искать пути получения из одних веществ другие, осуществляя их качественные превращения. На базе познания глубинных свойств различных веществ возникла теоретическая химия, которая в настоящее время представляет собой высокоупорядоченную и постоянно развивающуюся систему знаний. В наши дни химическая наука дает возможность получать вещества с заданными свойствами, находить способы управления этими свойствами, что является основной проблемой химии и системообразующим началом ее как науки.
1. История химии, алхимия
1.1 История алхимии
Алхимия складывалась в эпоху эллинизма на основе слияния прикладной химии египтян с греческой натурфилософией, мистикой и астрологией (золото соотносили с Солнцем, серебро – с Луной, медь – с Венерой, и т.д.) (II–VI вв.) в александрийской культурной традиции, представляя собой форму ритуально-магического (герметического) искусства. Алхимия – это самозабвенная попытка найти способ получения благородных металлов. Алхимики считали, что ртуть и сера разной чистоты, соединяясь в различных пропорциях, дают начало металлам, в том числе и благородным. В реализации алхимического рецепта предполагалось участие священных или мистических сил (частицы Бога или дьявола, надъестественного бытия, в котором проявления человеческого мира теряют свою силу), а средством обращения к этим силам было слово (заклинание, молитва) – необходимая сторона ритуала. Поэтому алхимический рецепт выступал одновременно и как действие, и как священнодействие.
В средневековой алхимии (ее расцвет пришелся на XIII–XV вв.) выделялись две тенденции. Первая – это мистифицированная алхимия, ориентированная на химические превращения (в частности, ртути в золото) и, в конечном счете, на доказательство возможности человеческими усилиями осуществлять космические превращения (давать человеку могущество над духами, воскрешать из мертвого (палингенезия) и, наконец, искусственно создать одушевленное существо – андроида или гомункула). В русле этой тенденции арабские алхимики сформулировали идею «философского камня» – гипотетического вещества, ускорявшего «созревание» золота в недрах земли; это вещество заодно трактовалось и как эликсир жизни, исцеляющий болезни и дающий бессмертие.
Вторая тенденция была больше ориентирована на конкретную практическую технохимию. В этой области достижения алхимии несомненны. К ним следует отнести: открытие способов получения серной, соляной, азотной кислот, селитры, сплавов ртути с металлами, многих лекарственных веществ, создание химической посуды и др.
Деятельность алхимика опиралась на некоторую совокупность «теоретических» представлений и образов. В их основе представление о том, что исходное материальное начало – первичная материя – хаотична, бесформенна и потенциально содержит в себе все тела, все минералы и металлы. Порожденные первоматерией тела уже не исчезают, но зато могут быть превращены друг в друга. Между первоматерией и отдельными порожденными ею материальными телами есть два промежуточных «звена».
Первое звено – всеобщие качественные начала – мужское («сера») и женское («ртуть» или «меркурий»); в XV в. к ним добавили еще одно начало – «соль» (движение). При этом следует иметь в виду, что такие «названия ни в коем случае нельзя смешивать с общеупотребительными, так «сера» в металлах обозначает цвет, горючесть, твердость, способность соединяться с другими металлами, тогда как «меркурий» значит блеск, летучесть, плавкость, ковкость. Что же касается «соли», то этим именем обозначали принцип, соединяющий «серу» с «меркурием», подобно жизненному началу, связывающему дух с телом»[1]. Второе звено – это состояния первоэлементов: земля (твердое состояние тела), огонь (лучистое состояние), вода (жидкое состояние), воздух (газообразное состояние), квинтэссенция (эфирное состояние). Алхимики полагали, что в результате взаимодействия качественных начал и состояний первоэлементов можно осуществлять любые трансмутации веществ.
Особое отношение к алхимии складывалось в системах светской и церковной власти. С одной стороны, крупные феодалы рассчитывали с помощью алхимии поправить свое материальное положение и потому преклонялись перед алхимией и ее «возможностями». С другой стороны, власть имущие к алхимии относились подозрительно. Так, римский император Диоклетиан в 296 г., опасаясь, что получение алхимиками золота ослабит его казну и экономику, приказал уничтожить все алхимические рукописи. По тем же причинам в 1317 г. папа Иоанн ХХП предал алхимию анафеме. Но это не помогло, и еще много столетий (вплоть до середины XVIII в.) алхимия оставалась элементом европейской духовной культуры.
1.2 История химии
Во второй половине XVII в. алхимическая традиция постепенно исчерпывает себя. Все более укреплялось представление о том, что существует некоторый предел, граница взаимопревращения веществ. Этот предел определяется составом химических веществ. В XVII–XVIII в. химия постепенно становится наукой о качественных изменениях тел, происходящих в результате изменения их состава.
Строгая дефиниция химии основывается на широком, вневременном универсальном смысле – как области естествознания и человеческой практики, связанной с химическими элементами и их комбинациями. Слово «химия» происходит либо от наименования Древнего Египта «Хем» («темный», «черный» – очевидно, по цвету почвы в долине реки Нил; смысл же названия – «египетская наука»), либо от древнегреч. «chemeia» – искусство выплавки металлов. Современное название химии производится от позднелат. «chimia» и является интернациональным, например нем. «Chemie», франц. «chimie», англ. «chemistry». Термин «Xимия» впервые употребил в 5 в. греч. алхимик Зосима.
Новому пониманию предмета химического познания способствовало возрождение античного атомизма. Здесь важную роль сыграли труды французского мыслителя П. Гассенди. Он не только воскресил атомистическую теорию, но, по словам Дж. Бернала, превратил её «в учение, куда вошло всё то новое в физике, что было найдено в эпоху Возрождения». Для обнаружения частиц, не видимых простым глазом, Гассенди использовал энгиоскоп (микроскоп); из этого он делал вывод, что если можно обнаружить столь мелкие частицы, то могут существовать и совсем мельчайшие, которые удастся увидеть впоследствии.
Он считал, что Бог создал определённое число атомов, отличающихся друг от друга формой, величиной и весом. Все в мире состоит из них. Как из кирпича, брёвен и досок можно построить огромное число разнообразных зданий, так и из нескольких десятков видов атомов природа создаёт великое множество тел. Соединяясь, атомы дают более крупные образования – «молекулы». Последние в свою очередь, объединяясь друг с другом, становятся более крупными и «доступными для ощущения». Тем самым Гассенди первым ввёл в химию понятие молекула (от латинского moles – с уменьшительным суффиксом cula).
И вместе с тем П. Гассенди, как и Р. Декарт, разделял заблуждения науки своего времени. Он признавал божественное происхождение атомов, признавал, что существуют особые атомы запаха, вкуса, тепла и холода.
Развитию корпускулярной теории способствовал великий английский учёный Исаак Ньютон (1643–1727), занимавшийся и вопросами химии. Он имел хорошо оборудованную химическую лабораторию, среди его трудов есть сочинение «О природе кислот» (1710). Ньютон считал, что корпускулы созданы Богом, что они неделимы, тверды и неуничтожимы. Соединение корпускул происходит за счёт притяжения, а не за счёт крючков, зазубрин и т.д. Такое притяжение и определяет «химическое действие»; распад существующих веществ на первичные частицы и образование из них других сочетаний обусловливают появления новых веществ.
Корпускулярное учение нашло свое завершение в трудах знаменитого английского учёного Роберта Бойля.
Бойль собрал богатую библиотеку и оборудовал прекрасную лабораторию, где работал со своими помощниками. Молодой учёный разработал основы анализа (от «анализис» – разложение) «мокрым путём», т.е. анализ в растворах. Он ввёл индикаторы (настой лакмуса, цветов фиалок, а также лакмусовые бумажки) для распознания кислот и щелочей; соляную кислоту и её соли с помощью нитрата серебра, соли серной кислоты – с помощью извести и т.д. Эти приёмы используются и сейчас.
Под влиянием работ Торчелли по изучению атмосферного давления Бойль занялся исследованием свойств воздуха. Он брал трубки U-образной формы с разной длиной колен. Короткое было запаяно, а длинное открыто. Заливая в последнее ртуть, Бойль «запирал» в коротком колене. Если изменять количество ртути в длинном колене, то изменяется объём воздуха в коротком. Так была установлена закономерность: объём газа обратно пропорционален его давлению (1662). Позднее эту закономерность наблюдал французский учёный Э. Мариотт. Сейчас первый газовый закон именуется законом Бойля – Мариотта.
А за год до открытия газового закона Бойль опубликовал книгу «Химик-скептик», в которой изложил свои взгляды. Бойль считает химию самостоятельной наукой, а не подспорьем алхимии и медицины. Все тела, пишет он, состоят из движущихся частиц, обладающих разной величиной и формой, а элементами, подчёркивает Бойль, не могут быть ни «начала» Аристотеля, ни «начала» алхимиков. Это «определённые, первоначальные и простые, вполне несмешанные тела, которые не составлены друг из друга, но представляют собой те составные части, из которых составлены все так называемые смешанные тела и которые они, в конце концов, могут быть разложены».
Таким образом, элементы, по Бойлю, это вещества, которые нельзя разложить (т.е. простые вещества), они состоят из однородных корпускул. Таковы золото, серебро, олово, свинец. Другие, например, киноварь, разлагающиеся на ртуть и серу, он относил к сложным веществам. В свою очередь, серу, ртуть, которые не удалось разложить, следовало отнести к элементам. Ему удалось в корпускулярной теории строения веществ объединить два подхода – учение об элементах и атомистические представления. «Бойль делает из химии науку» – писал Ф. Энгельс.
2. Неорганическая и органическая химия
2.1 Неорганическая химия
Неорганическая химия, наука о хим. элементах и образуемых ими простых и сложных веществах, за исключением органических соединений.
Понятие «неорганическая химия» (минер. химия) появилось первоначально для обозначения веществ минерального происхождения.
Основные задачи современной неорганической химии: изучение строения, свойств и химических реакций простых веществ и соединений, взаимосвязи строения со свойствами и реакционной способностью веществ, разработка методов синтеза и глубокой очистки веществ, общих методов получения неорганических материалов.
По изучаемым объектам неорг. химию подразделяют на химию отдельных элементов, химию групп элементов в составе периодичной системы (химия щелочных металлов, щелочноземельных элементов, галогенов, халькогенов и др.), химию определенных соединений тех или иных элементов (химия силикатов, пероксидных соединений и др.), химию элементов, объединенных в группы по исторически сложившимся признакам (напр., химия редких элементов), химию близких по свойствам и применению веществ (химия тугоплавких веществ, интерметаллидов, полупроводников, энергонасыщенных соединений, благородных металлов, неорг. полимеров и др.). Нередко обособляют химию переходных элементов.
Как и многие др. хим. науки, неорганическая химия неразрывно связана с физ. химией, которая может считаться теоретической и методологической основой современной химии, с аналитической химией – одним из главных инструментов химии.
Неорг. химия отчасти пересекается с орг. химией, особенно с химией металлоорганических соединении, бионеорганической химией и др.
Теоретические представления неорг. химии используют в геохимии, космохимии, химии твердого тела, химии высоких энергий, радиохимии, ядерной химии, в некоторых разделах биохимии и агрохимии.
Прикладная часть неорг. химии связана с хим. технологией, металлургией, галургией, электроникой, с добычей полезных ископаемых, производством керамики, строительных, конструкционных и др. неорг. материалов, с обеспечением работы энергетических установок (например, АЭС), с сельским хозяйством, с обезвреживанием промышленных отходов, охраной природы и др.
История неорг. химии тесно связана с общей историей химии, а вместе с ней – с историей естествознания и историей человеческой цивилизации.
Этапными для развития неорг. химии явились работы И. Берцелиуса, который в 1814 опубликовал таблицу атомных масс. А. Авогадро и Ж. Гей-Люссак открыли газовые законы, П. Дюлонг и А. Пти нашли правило, связывающее теплоемкость с числом атомов в соединении, Г.И. Гесс – закон постоянства количества теплоты. Возникла атомно-молекулярная теория.
В 1807 Г. Дэви электрохимически разложил гидроксиды натрия и калия и ввел в практику новый метод выделения простых веществ; в 1834 М. Фарадей опубликовал основные законы электрохимии.
2-я половина – конец XIX в. ознаменовались обособлением физ. химии. К. Гульдберг и П. Вааге сформулировали закон действующих масс. Работы С. Аррениуса, Я. Вант-Гоффа, В. Оствальда положили начало теории растворов.
В этот же период зародилось учение о валентности (Ф. Кекуле, Ш. Вюрц и др.), стали известными новые хим. элементы (бор, литий, кадмий, селен, кремний, бром, алюминий, йод, торий, ванадий, лантан, эрбий, тербий, диспрозий, рутений, ниобий), с помощью введенного в практику спектрального анализа было доказано существование цезия, рубидия, таллия и индия. Было проведено определение и уточнение атомных масс многих химических элементов.
К кон. 1860-х гг. стало известно 63 хим. элемента и большое число разнообразных хим. соединений, однако научная классификация элементов отсутствовала. Основой для систематики явился периодический закон Менделеева, с помощью которого были исправлены атомные массы многих элементов и предсказаны свойства неизвестных в то время веществ. Последние открытия Галлия (П.Э. Лекок де Буабодран, 1875), Скандия (Л. Нильсон, 1879), Германия (К.А. Винклер, 1886), Лантаноидов, благородных газов (У. Рамзай, 1894–98), первых радиоактивных элементов – полония и радия (М. Склодовская, П. Кюри, 1898) блестяще подтвердили периодический закон. При получении астата, актиноидов, курчатовия, нильсбория и элементов с атомными номерами 106 и выше этот закон был использован на практике. Приоритет Менделеева в открытии периодического закона, некоторое время оспаривавшийся Л. Мейером, был закреплен в названии одного из искусственных элементов (менделевия).
Теория строения атома (Э. Резерфорд, 1911; Н. Бор, 1913), введение понятия атомного номера (Г. Мозли, 1914) позволили дать периодическому закону физическое обоснование.
Теоретическая неорганическая химия. Этот раздел неорг. химии рассматривает вопросы хим. связи в неорг. веществах, структуры веществ, их свойства и реакционная способность. Основными в неорг. химии являются периодический закон, закон постоянства состава веществ и др. Однако ключевой проблемой сейчас является природа хим. связи. В неорг. веществах встречаются все виды хим. связи – ковалентная, ионная и металлическая. Теория хим. связи, в частности, рассматривает вопросы природы связи, ее энергии, длины, полярности. Наибольшее распространение получили методы молекулярных орбиталей, наряду с которыми используют метод валентных связей, теорию кристаллического поля и др. Для неорг. химии особенно актуально приложение методов молекулярных орбиталей к твердым телам.
Большое значение придается спектрам в электромагнитном диапазоне (для определения структуры веществ) и магнитным свойствам веществ (в целях создания магнитных материалов). Теоретическая неорг. химия активно использует методы хим. термодинамики и хим. кинетики.
Теоретическая неорг. химия изучает также закономерности образования дефектов кристаллической решетки, влияние дефектов на свойства веществ, исследует кинетику твердофазных процессов.
Некоторые вопросы, являются одновременно и проблемами физики и физ. химии. Например, квантово-химическое описание электронной конфигурации атомов и ионов, проблемы происхождения хим. элементов и их превращений в космосе, создание теории высокотемпературной сверхпроводимости и др.
Прикладная химия. Еще в 18 в. установилась тесная связь между неорганической химией и ремеслами – основой зарождавшейся промышленности. Позднее неорг. химия стала научной базой многих производств, определяющих уровень промышленного развития отдельных стран и всего человечества.
Прикладной частью неорг. химии традиционно считается технология неорг. веществ. Она связана с крупномасштабными производствами серной, соляной, фосфорной, азотной кислот, соды, аммиака, хлора, фтора, фосфора, а также солей натрия, калия, магния и др., диоксида углерода, водорода, различных минеральных удобрений и мн. др. веществ. Большая часть этих продуктов потребляется др. химическими производствами и металлургией.
Прикладная неорг. химия играет существенную роль в развитии важнейших отраслей народного хозяйства. Так, в машиностроении и строительстве широко используют материалы, получаемые из минерального сырья хим. методами. Это, например, металлы и сплавы, минеральные красители, твердые сплавы для режущего инструмента.
2.2 Органическая химия
Органическая химия, наука, изучающая соединения углерода с другими элементами (органические соединения), а также законы их превращений. Название «органическая химия» возникло на ранней стадии развития науки, когда предмет изучения ограничивался соединениями углерода растительного и животного происхождения. Не все соединения углерода классифицируются как органические. Например, СО2, HCN, CS2 традиционно относят к неорг. Условно можно считать, что прототипом орг. соединений является метан СН4.
К настоящему времени число известных орг. соединений превышает 10 млн. и увеличивается каждый год на 250–300 тыс. Многообразие орг. соединений определяется уникальной способностью атомов углерода соединяться друг с другом простыми и кратными связями, образовывать соединения с практически неограниченным числом атомов, связанных в цепи, циклы, бициклы, трициклы, полициклы, каркасы и др., образовывать прочные связи почти со всеми элементами периодичной системы, а также явлением изомерии – существованием разных по свойствам веществ, обладающих одним и тем же составом и молярной массой.
Многообразие и громадное число орг. соединений определяет значение орг. химии как крупнейшего раздела современной химии. Окружающий нас мир построен главным образом из орг. соединений; пища, топливо, одежда, лекарства, краски, моющие средства, материалы, без которых невозможно создание транспорта, книгопечатания, проникновение в космос и прочее, – все это состоит из орг. соединений. Важнейшую роль орг. соединения играют в процессах жизнедеятельности. Отдельный раздел орг. химии составляет химия высокомолярных соединений: по величине молекул орг. вещества делятся на низкомолекулярные (с молярной массой от нескольких десятков до нескольких сотен, редко до тысячи) и высокомолекулярные (макромолекулярные; с молярной массой порядка 104-106 и более).
Орг. химия изучает не только соединения, получаемые из растительных и животных организмов, но в основном соединения, созданные искусственно с помощью лаборатории или промышленного органического синтеза. Более того, объектами изучения компьютерной орг. химии являются соединения, не только не существующие в живых организмах, но которые, по-видимому, нельзя получить искусственно (напр., гипотетический аналог метана, имеющий не природное тетраэдрич. строение, а форму плоского квадрата, в центре которого лежит атом С, а в вершинах – атомы Н).
Классификация органических соединений. Основу орг. соединений составляет незамкнутая (открытая) или замкнутая цепь углеродных атомов; одно или несколько звеньев цепи может быть заменено на атомы, отличные от углерода, – гетероатомы, чаще всего О, N, S. По структуре орг. соед. подразделяют на алифатические соединения – углеводороды и их производные, имеющие открытую углеродную цепь; карбоциклические соединения с замкнутой углеродной цепью; гетероциклические соединения. Углеводороды и их производные, не содержащие кратных связей, относятся к насыщенным соединениям, с кратными связями – к ненасыщенным.
Историческая справка. Истоки орг. химии восходят к глубокой древности (уже тогда знали о спиртовом и уксуснокислом брожении, крашении индиго и ализарином). Однако в средние века (период алхимии) были известны лишь немногие индивидуальные орг. вещества. Все исследования этого периода сводились главным образом к операциям, при помощи которых, как тогда думали, одни простые вещества можно превратить в другие. Начиная с ХVI в. (период ятрохимии) исследования были направлены в основном на выделение и использование различных лекарственных веществ: был выделен из растений ряд эфирных масел, приготовлен диэтиловый эфир, сухой перегонкой древесины получены древесный (метиловый) спирт и уксусная кислота, из винного камня – винная кислота, перегонкой свинцового сахара – уксусная кислота, перегонкой янтаря – янтарная.
Слияние хим. соединений растительного и животного происхождения в единую хим. науку орг. химии осуществил Й. Берцелиус, который ввел сам термин и понятие орг. вещества, образование последнего, по Берцелиусу, возможно только в живом организме при наличии «жизненной силы». Это заблуждение опровергли Ф. Вёлер (1828), который получил мочевину (орг. вещество) из цианата аммония (неорг. вещество), А. Кольбе, синтезировавший уксусную кислоту, М. Бертло, получивший метан из H2S и CS2, A.M. Бутлеров, синтезировавший сахаристые вещества из формалина. В первой пол. XIX в. был накоплен обширный опытный материал и сделаны первые обобщения, определившие бурное развитие орг. химии: развиты методы анализа орг. соединения (Берцелиус, Ю. Либих, Ж. Дюма, М. Шеврёль), создана теория радикалов (Вёлер, Ж. Гей-Люссак, Либих, Дюма) как групп атомов, переходящих неизменными из исходной молекулы в конечную в процессе реакции; теория типов (Ш. Жерар, 1853), в которой орг. соединения конструировались из неорг. веществ – «типов» замещением в них атомов на орг. фрагменты; введено понятие изомерии (Берцелиус).
Одновременно продолжается интенсивное развитие синтеза. Создаются первые промышленные производства орг. соед. (А. Гофман, У. Перкин-старший – синтетич. красители: мовеин, фуксин, цианиновые и азокрасители). Усовершенствование открытого Н.Н. Зининым (1842) способа синтеза анилина послужило основой создания анилинокрасочной промышленности.
Идея неразрывной связи хим. и физ. свойств молекулы с ее строением, идея единственности этого строения впервые была высказана Бутлеровым (1861), который создал классическую теорию хим. строения (атомы в молекулах соединяются согласно их валентностям, хим. и физ. свойства соединения определяются природой и числом входящих в их состав атомов, а также типом связей и взаимным влиянием непосредственно несвязанных атомов). Теория хим. строения определила дальнейшее бурное развитие орг. химии: в 1865 Кекуле предложил формулу бензола, позднее высказал идею об осцилляции связей; В.В. Марковников и А.М. Зайцев сформулировали ряд правил, впервые связавших направление хим. реакции с хим. строением вступающего в реакцию вещества.
Работами Байера, К. Лаара, Л. Клайзена, Л. Кнорра развиты представления о таутомерии – подвижной изомерии. Все эти теоретические представления способствовали мощному развитию синтетической химии. К кон. XIX в. были получены все важнейшие представители углеводородов, спиртов, альдегидов и кетонов, карбоновых кислот, галогено- и нитропроизводных, азот- и серосодержащих структур, гетероциклов ароматической природы. Разработаны методы получения диенов, ацетиленов и алленов (А.Е. Фаворский). Открыты многочисленные реакции конденсации (Ш. Вюрц, А.П. Бородин, У. Перкин, Клайзен, А. Михаэль, Ш. Фридель, Дж. Крафтс, Э. Кнёвенагель и др.). Исключительные успехи были достигнуты Э.Г. Фишером в изучении углеводов, белков и пуринов, в использовании ферментов в орг. синтезе (1894), им же был осуществлен синтез полипептидов. Основой промышленности душистых веществ становятся работы О. Валлаха по химии терпенов. Выдающимися даже для нашего времени являются пионерские работы Р. Вильштеттера. Фундаментальный вклад в развитие орг. синтеза был внесен В. Гриньяром (1900–20) и Н.Д. Зелинским (1910) – создание исключительно плодотворного метода синтеза магнийорганических соединений и открытие каталитических превращений углеводородов; последнее сыграло выдающуюся роль в развитии химии нефти. Химия свободных радикалов началась с работ М. Гомберга (1900), открывшим трифенилметильный радикал, и была продолжена работами А.Е. Чичибабина, Г. Виланда и Ш. Гольдшмидта.
Строение органических соединений. Для орг. соединений характерны неполярные ковалентные связи С–С и полярные ковалентные связи С–О, С–N, С–Hal, С–металл и т.д. Образование ковалентных связей было объяснено на основании развитых Г. Льюисом и В. Косселем (1916) предположений о важной роли электронных образований – октетов и дублетов. Молекула устойчива, если валентная оболочка таких элементов, как С, N, О, Hal, содержит 8 электронов (правило октета), а валентная оболочка водорода – 2 электрона. Хим. связь образуется обобществленной парой электронов различных атомов (простая связь). Двойные и тройные связи образуются соотвующимися двумя и тремя такими парами. Электроотрицательные атомы (F, О, N) используют для связи с углеродом не все свои валентные электроны; «неиспользованные» электроны образуют неподеленные (свободные) электронные пары. Полярность и поляризуемость ковалентных связей в орг. соединениях в электронной теории Льюиса – Косселя объясняется смещением электронных пар от менее электроотрицательного к более электроотрицательному атому, что находит выражение в индуктивном эффекте и мезомерном эффекте.
Классическая теория хим. строения и первоначально электронные представления оказались не в состоянии удовлетворительно описать на языке структурных формул строение многих соединений, например, ароматических. Современная теория связи в орг. соединениях основана главным образом на понятии орбиталей и использует методы молекулярных орбиталей. Интенсивно развиваются квантовохим. методы, объективность которых определяется тем, что в их основе лежит аппарат квантовой механики, единственно пригодный для изучения явлений микромира.
Общая характеристика реакций органических соединений. Реакции орг. соединений имеют некоторые специфические особенности. В реакциях неорг. соединений обычно участвуют ионы; эти реакции протекают очень быстро, иногда мгновенно при нормальной температуре. В реакциях орг. соединений обычно участвуют молекулы; при этом одни ковалентные связи разрываются, а другие образуются. Такие реакции протекают медленнее ионных, и для их ускорения часто требуется повысить температуру или добавить катализатор. Наиболее часто используют в качестве катализаторов и основания. Обычно протекает не одна, а несколько реакций, так что выход нужного продукта очень часто составляет менее 50%.
Возникновение органических соединений. Большинство орг. соединений в природе образуется в процессе фотосинтеза из диоксида углерода и воды под действием солнечного излучения, поглощаемого хлорофиллом в зеленых растениях. Однако орг. соединений должны были существовать на земле и до возникновения жизни, которая не могла появиться без них. Первичная земная атмосфера около 2 млрд. лет назад имела восстановительные свойства, т.к. в ней не было кислорода, а содержались прежде всего водород и вода, а также СО, азот, аммиак и метан.
В
условиях сильного радиоактивного излучения земных минералов и интенсивных
атмосферных разрядов в атмосфере протекал абиотический синтез аминокислот по
схеме: CH4 + H2O + NH3![]() Аминокислоты.
Аминокислоты.
Возможность такой реакции в настоящее время доказана лабораторными опытами.
3. Реакционная способность веществ, анализ и синтез
3.1 Реакционная способность веществ
Реакционная способность, характеристика относительной хим. активности молекул, атомов, ионов, радикалов. Для количественной оценки рассматривают реакционные серии, т.е. ряды однотипных реакций, проводимых в одинаковых условиях, например: (стандартная реакция)
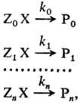
где Х – общая группа атомов, которая претерпевает изменения в данной реакции (реакционный центр), Z0, Zl,…, Zn – неизменяющиеся молекулярные фрагменты, Р0, Р1,…, Рn – продукты реакции. Отношения констант скоростей k1/k0,…, kn/k0 количественно характеризуют реакционную способность. В ряду реагентов ZiX (i = 0, 1,…, п). В правильно составленной реакции серии изменение механизма реакции должно быть исключено, т.е. константы скорости должны характеризовать одну и ту же элементарную реакцию.
Типичные реакционные серии. Простейшая ситуация возникает при анализе изомерного состава продуктов реакции. В реакции замещения в ароматическом ряду в зависимости от заместителя R образуются те или иные изомеры, например, при нитровании:
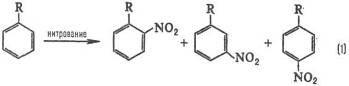
Электронодонорные заместители [R=СН3, ОСН3, N(CH3)2] стимулируют образование орто- и пара-продуктов, а электроноакцепторные (R = СООН, SO3H, NO2) – мета-продуктов, причем в первом случае реакция идет легче, чем с незамещенным бензолом (R = Н), а во втором – труднее. Эти закономерности называются правилами ориентации в ароматическом ряду. Стереохимическая направленность перипиклической реакций определяется Вудворда-Хофмаина правилами, например:
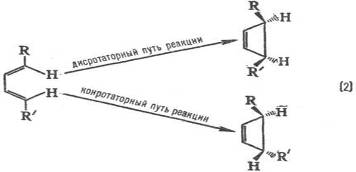
При дисротаторном пути реакции заместители R и R' в продукте будут расположены по одну сторону плоскости цикла, при конротаторном пути – по разные стороны. Эксперимент показывает, что термическая циклизация производных бутадиена происходит по конротаторному пути, а фотохимическая циклизация – по дисротаторному пути.
В примерах (1) и (2) нет необходимости в количественных кинетических измерениях, реакционная способность определяется по относительному выходу изомеров. Пример широкой реакционной серии – реакции радикального присоединения по двойной связи:
![]()
Реакционная способность характеризуется отношением константы скорости k к константе скорости k0 реакции с этиленом (R, R' = Н) (см. табл.). Аналогичные кинетические измерения сделаны для реакций присоединения метильного радикала к ароматическим молекулам и для реакций присоединения др. радикалов.

Квантовохимическая теория реакционной способности. Современная теоретическая химия позволяет непосредственно рассчитать константы скорости только для несложных хим. систем. В теории реакционной способности качественной закономерности могут быть выявлены для объектов любой сложности. При этом используют различные подходы. При эмпирическом подходе классифицируют влияние заместителей по нескольким типам (эффекты сопряжения, полярные, пространственные и др.) и применяют корреляционные соотношения. Традиционный квантовохимический подход основан на активированного комплекса теории; при этом предполагается, что для всех реакций, составляющих реакционную серию, остается примерно постоянным множитель А в Аррениуса уравнении для константы скорости k=Aexp(-E./RT). Поэтому характеристикой реакционной способности служит энергия активации реакции E., которая практически совпадает с высотой потенциального барьера на поверхности потенциальной энергии (ППЭ).
Советскому ученому Н.Н. Семенову предстояло открыть разветвленные цепные реакции. Теория разветвленных цепных реакций дала начало новому направлению исследований – химической физике, дисциплине, промежуточной между физикой и химией.
В химии были также открыты колебательные реакции, получившие название «химических часов». Основа колебательной реакции – наличие двух типов молекул, способных превращаться друг в друга. Назовем один из них А (красные молекулы), другой – В (синие). Мы привыкли думать, что химическая реакция – это хаотические, происходящие наобум столкновения частиц. По этой логике взаимные превращения А и В должны приводить к усредненному цвету раствора со случайными вспышками красного и синего. Но когда условия далеки от равновесных, происходит совершенно иное: раствор в целом становится красным, потом синим, потом снова красным. Получается, будто молекулы как бы устанавливают связь между собой на больших, макроскопических расстояниях через большие, макроскопические отрезки времени. Появляется нечто похожее на сигнал, по которому все А или все В реагируют разом… Такое поведение традиционно приписывалось только живому – теперь же ясно, что оно возможно и у систем сравнительно простых, неживых.
3.2 Анализ
Молекулярный анализ, установление качественного и количественного состава хим. соединений и их смесей.
При качественном анализе смеси хим. соединений обычно предварительно разделяют различными методами (хроматографией, ректификацией, кристаллизацией, экстракцией, осаждением, термической диффузией и др.); затем для разделенных веществ определяют так называемые интегральные молекулярные признаки, к которым относятся молярная масса, суммарный элементный состав, плотность, растворимость, показатели преломления, потенциалы ионизации, а также спектры поглощения электромагнитного излучения, масс-спектры и т.п. Эти характеристики хим. соединений сопоставляют с соответствующими константами и спектрами образцов сравнения, устанавливают отсутствие депрессии (понижение и увеличение интервала) температуры плавления смеси идентифицируемого соединения и эталонного вещества (т.е. известного вещества, отождествляемого с исследуемым). Часто определяют хроматографической характеристики веществ (индексы удерживания, объемы удерживания и др.); при этом одновременно идентифицируемое вещество отделяется от других компонентов смеси. Идентификацию можно считать достоверной только в том случае, если совпадают несколько характеристик и констант идентифицируемого и эталонного веществ. Наиболее эффективны комбинированные методы: хромато-масс-спектрометрия, сочетание высокоэффективной жидкостной хроматографии и масс-спектрометрии, сочетание газо-жидкостной или жидкостной хроматографии и др. При качественном молекулярном анализе смеси веществ без предварительного разделения хим. соединения обнаруживают по характерным хим. реакциям, спектрам поглощения, масс-спектрами и т.п.
Иногда под молекулярным анализом понимают только установление строения хим. соединений. При этом сначала определяют его эмпирическую формулу по данным качественного и количественного элементного анализа.
3.3 Органический синтез
Органический синтез имеет важнейшее практическое значение. На протяжении ХХ в. было синтезировано огромное количество веществ, которые до этого человечество находило лишь в природном состоянии – различные лекарства, витамины, удобрения, детергенты, каучук и т.д. В настоящее время ведутся работы по выработке технологии создания материалов не только из неорганических соединений, а из растительного сырья: кукурузы (из ее стеблей, которые сжигают) и т.п. Одно из перспективных направлений – создание биодеградируемой упаковки. «Представьте, баночка из-под йогурта, брошенная не очень-то культурным человеком в воду или на газон, в считанные дни исчезнет, разложившись до углекислого газа и воды» («Химия и жизнь», 2001, №12. С. 17), и количество углекислого газа при этом не увеличивается, как при использовании нефти. Это получило название «зеленая химия».
Химия идет по технологическому пути, так как свойства естественных молекул уже достаточно хорошо изучены, и задача – создавать новые вещества с новыми, неизвестными природе свойствами, как, например, пластмассы. Ежегодно синтезируется более 6000 новых химических соединений. И необходимо включать их в природные кругообороты, чтобы не осложнялись экологические проблемы.
С появлением новых промышленных процессов, средств связи (например, сотовой связи) возникает все большая потребность в новых материалах. Необычное и даже неожиданное словосочетание – интеллектуальные, или разумные, материалы – вещества нового поколения, которые оптимизируют свои характеристики в зависимости от внешних условий. Такие материалы откликаются на всякое физическое воздействие: крыло новой машины автоматически меняет свою форму, чтобы оптимально соответствовать аэродинамическим условиям или оптимизировать угол атаки. Некоторые детали (например, лопатки турбин) выращивают из расплава как кристалл – целиком нужной формы. Такова химия XXI в.
4. Важнейшие химические связи и методы исследования веществ
Как известно, атомы могут соединяться друг с другом с образованием как простых, так и сложных веществ. При этом образуются различного типа химические связи: ионная, ковалентная (неполярная и полярная), металлическая и водородная. Одно из наиболее существенных свойств атомов элементов, определяющих, какая связь образуется между ними – ионная или ковалентная, – это электроотрицательность, т.е. способность атомов в соединении притягивать к себе электроны.
Условную количественную оценку электроотрицательности дает шкала относительных электроотрицательностей.
В периодах наблюдается общая тенденция роста электроотрицательности элементов, а в группах – их падения. Элементы по электроотрицательностям располагают в ряд, на основании которого можно сравнить электроотрицательности элементов, находящихся в разных периодах.
Тип химической связи зависит от того, насколько велика разность значений электроотрицательностей соединяющихся атомов элементов. Чем больше отличаются по электроотрицательности атомы элементов, образующих связь, тем химическая связь полярнее. Провести резкую границу между типами химических связей нельзя. В большинстве соединений тип химической связи оказывается промежуточным; например, сильнополярная ковалентная химическая связь близка к ионной связи. В зависимости от того, к какому из предельных случаев ближе по своему характеру химическая связь, ее относят либо к ионной, либо к ковалентной полярной связи.
Ионная связь образуется при взаимодействии атомов, которые резко отличаются друг от друга по электроотрицательности. Например, типичные металлы литий(Li), натрий(Na), калий(K), кальций (Ca), стронций(Sr), барий(Ba) образуют ионную связь с типичными неметаллами, в основном с галогенами.
Кроме галогенидов щелочных металлов, ионная связь также образуется в таких соединениях, как щелочи и соли. Например, в гидроксиде натрия(NaOH) и сульфате натрия(Na2SO4) ионные связи существуют только между атомами натрия и кислорода (остальные связи – ковалентные полярные).
Ковалентная неполярная связь
При взаимодействии атомов с одинаковой электроотрицательностью образуются молекулы с ковалентной неполярной связью. Такая связь существует в молекулах следующих простых веществ: H2, F2, Cl2, O2, N2. Химические связи в этих газах образованы посредством общих электронных пар, т.е. при перекрывании соответствующих электронных облаков, обусловленном электронно-ядерным взаимодействием, которые осуществляет при сближении атомов.
Составляя электронные формулы веществ, следует помнить, что каждая общая электронная пара – это условное изображение повышенной электронной плотности, возникающей в результате перекрывания соответствующих электронных облаков.
Ковалентная полярная связь
При взаимодействии атомов, значение электроотрецательностей которых отличаются, но не резко, происходит смещение общей электронной пары к более электроотрицательному атому. Это наиболее распространенный тип химической связи, которой встречается как в неорганических, так и органических соединениях.
К ковалентным связям в полной мере относятся и те связи, которые образованы по донорно-акцепторному механизму, например в ионах гидроксония и амония.
Металлическая связь
Связь, которая образуется в результате взаимодействия относительно свободных электронов с ионами металлов, называются металлической связью. Этот тип связи характерен для простых веществ – металлов.
Сущность процесса образования металлической связи состоит в следующем: атомы металлов легко отдают валентные электроны и превращаются в положительные заряженные ионы. Относительно свободные электроны, оторвавшиеся от атома, перемещаются между положительными ионами металлов. Между ними возникает металлическая связь, т.е. Электроны как бы цементируют положительные ионы кристаллической решетки металлов.
Водородная связь
Связь, которая образуется между атомов водорода одной молекулы и атомом сильно электроотрицательного элемента (O, N, F) другой молекулы, называется водородной связью.
Может возникнуть вопрос: почему именно водород образует такую специфическую химическую связь?
Это объясняется тем, что атомный радиус водорода очень мал. Кроме того, при смещении или полной отдаче своего единственного электрона водород приобретает сравнительно высокий положительный заряд, за счет которого водород одной молекулы взаимодействует с атомами электроотрицательных элементов, имеющих частичный отрицательный заряд, выходящий в состав других молекул (HF, H2O, NH3).
Рассмотрим некоторые примеры. Обычно мы изображаем состав воды химической формулой H2O. Однако это не совсем точно. Правильнее было бы состав воды обозначать формулой (H2O) n, где n = 2,3,4 и т.д. Это объясняется тем, что отдельные молекулы воды связаны между собой посредством водородных связей.
Водородную связь принято обозначать точками. Она гораздо более слабая, чем ионная или ковалентная связь, но более сильная, чем обычное межмолекулярное взаимодействие.
Наличие водородных связей объясняет увеличения объема воды при понижении температуры. Это связано с тем, что при понижении температуры происходит укрепление молекул и поэтому уменьшается плотность их «упаковки».
При изучении органической химии возникал и такой вопрос: почему температуры кипения спиртов гораздо выше, чем соответствующих углеводородов? Объясняется это тем, что между молекулами спиртов тоже образуются водородные связи.
Повышение температуры кипения спиртов происходит также вследствие укрупнения их молекул.
Водородная связь характерна и для многих других органических соединений (фенолов, карбоновых кислот и др.). Из курсов органической химии и общей биологии вам известно, что наличием водородной связи объясняется вторичная структура белков, строение двойной спирали ДНК, т.е. явление комплиментарности.
5. Условия существования жизни, связи и функции живого вещества
Условия существования живого вещества. Жизнь, ее происхождение и развитие всегда, с древнейших времен волновали мысль человека.
Каковы же условия этой внешней среды обитания, благоприятствующие возникновению, сохранению и развитию жизни?
Экология рассматривает пять таких условий, совокупность которых определяет эти возможности (Мамедов и Суравегина, 1966).
1. Достаточное количество кислорода и углекислого газа. С появлением озонового экрана резко интенсифицируется освоение континентов лесной растительностью с ее бурным фотосинтезом и в относительно короткий срок, в несколько миллионов лет содержание свободного кислорода достигло современного уровня. Стоит отметить, что в настоящее время всеми растениями мира продуцируется около 100–150 млрд. т. кислорода, примерно в равных долях на суше и в океанических пространствах. Углекислый газ первоначально попадал в атмосферу и гидросферу земли из земных недр, с продуктами дегазации мантии, а затем – с вулканическими газами. Можно представить каталитическую реакцию графита с водой, происходящую при высоких температурах или температурную диссоциацию первичных карбонатов. Одновременно, удаление углекислого газа из атмосферы происходило и происходит главным образом за счет образования карбонатного скелета морских организмов и накопления на морском дне карбонатных отложений и органического вещества растений.
2. Достаточное количество жидкой воды, недостаток которой встречается на земле лишь в Антарктиде, Гренландии, высокогорьях и экваториальных пустынях. В момент образования нашей планеты из протопланетного облака все элементы ее будущей гидросферы, как и атмосферы, находились в составе твердых веществ (Монин, 1977): вода в гидроокислах, азот – в нитридах, кислород в окислах металлов, углерод – в графитах, карбидах и карбонатах. Возникновение гидросферы связано уже с последующими выплавками базальтов, водяного пара и газов, в первые 0,5–1,5 млрд. лет существования Земли, в результате прогревания недр планеты за счет гравитационного сжатия и распада радиоактивных изотопов. Подтверждением тому служат современные вулканические газы, содержащие водяной пар в количествах не менее 70–80 весовых процентов.
3. Определенный интервал благоприятных температур: не слишком низких для протекания биохимических реакций с участием ферментов и не слишком высоких, не более 1000°С, выше которых белок свертываются.
4. Необходимый минимум минеральных веществ в почвенном слое, доступных для освоения микроорганизмами и растениями.
5. Ограничение солености среды: при концентрации солей примерно в 10 раз выше, чем морская вода, а ее соленость составляет в среднем 35 г./кг, жизнь в воде исчезает, подземные же воды лишены жизни при их минерализации свыше 270 г./л.
6. Отсутствие загрязняющих веществ, которые по своим свойствам и концентрации превышают допустимые для биосферных объектов уровни. Таковы, меняющиеся под влиянием человека, необходимые условия существования живого вещества Земли.
Связи и функции веществ. Понятие «живое вещество» обозначает совокупность живых организмов биосферы. Область распространения включает нижнюю часть воздушной оболочки (атмосферы), всю водную оболочку (гидросферу), и верхнюю часть твёрдой оболочки (литосферы). В.И. Вернадским отметил, что между косной, безжизненной частью биосферы, косными природными телами и живыми организмами, её населяющими идёт непрерывный обмен энергией. Живое вещество играет наиболее важную роль по сравнению с другими веществами биосферы, и выполняет ряд важнейших функций.
Энергетическая функция выполняется, прежде всего, растениями, которые в процессе фотосинтеза аккумулируют солнечную энергию в виде разнообразных органических соединений. Чтобы биосфера могла существовать и развиваться, ей необходима энергия. Собственных источников энергии она не имеет и может потреблять энергию только от внешних источников. Главным источником для биосферы является Солнце. Солнечный свет для биосферы является рассеянной лучистой энергией электромагнитной природы. Почти 99% этой энергии, поступившей в биосферу, поглощается атмосферой, гидросферой и литосферой, а также участвует в вызванных ею физических и химических процессах (движение воздуха и воды, выветривание и др.) Только около 1% накапливается на первичном звене ее поглощения и передается потребителям уже в концентрированном виде. По словам Вернадского, зеленые хлорофилльные организмы, зеленые растения, являются главным механизмом биосферы
Каждый последующий этап развития жизни сопровождался все более интенсивным поглощением биосферой солнечной энергии. Одновременно нарастала энергоемкость жизнедеятельности организмов в изменяющейся природной среде, и всегда накопление и передачу энергии осуществляло живое вещество.
В отличие от зеленых растений некоторые группы бактерий синтезируют
органическое вещество за счет не солнечной энергии, а энергии, выделяющейся в процессе реакций окисления серных и азотных соединений. Этот процесс именуется хемосинтезом. В накоплении органического вещества в биосфере он, по сравнению с фотосинтезом, играет ничтожно малую роль.
Минерализация органических веществ, разложение отмершей органики до простых неорганических соединений, химическое разложение горных пород, вовлечение образовавшихся минералов в биотический круговорот определяет деструктивную (разрушительную) функцию живого вещества. Данную функцию в основном выполняют грибы, бактерии.
Мертвое органическое вещество разлагается до простых неорганических соединений (углекислого газа, воды, сероводорода, метана, аммиака и т.д.), которые вновь используются в начальном звене круговорота. Этим занимается специальная группа организмов – редуценты (деструкторы).
Особо следует сказать о химическом разложении горных пород. Благодаря живому веществу биотический круговорот пополняется минералами, высвобождаемыми из литосферы. Например, плесневый грибок в лабораторных условиях за неделю высвобождал из вулканической горной породы 3% содержащегося в ней кремния, 11% алюминия, 59% магния, 64% железа.
Концентрационная (накопительная) функция – избирательное накопление определенных веществ, рассеянных в природе – водорода, углерода, азота, кислорода, кальция, магния, натрия, калия, фосфора и многих других, включая тяжелые металлы, в живых существах. Раковины моллюсков, панцири диатомовых водорослей, скелеты животных – все это примеры проявления концентрационной функции живого вещества.
Способность концентрировать элементы из разбавленных растворов – это характерная особенность живого вещества. Наиболее активными концентраторами многих элементов являются микроорганизмы. Например, в продуктах жизнедеятельности некоторых из них по сравнению с природной средой содержание марганца увеличено в 1200000 раз, железа – в 65000, ванадия – в 420000, серебра – в 240000 раз.
Для построения своих скелетов или покровов активно концентрируют рассеянные минералы морские организмы.
Особого внимания заслуживает способность морских организмов накапливать микроэлементы, тяжелые металлы, в том числе ядовитые (ртуть, свинец, мышьяк), радиоактивные элементы. В теле беспозвоночных и рыб их концентрация может в сотни тысяч раз превосходить содержание в морской воде. Вследствие этого морские организмы полезны как источник микроэлементов, но вместе с тем употребление их в пищу может грозить отравлением тяжелыми металлами или быть опасным в связи с повышенной радиоактивностью.
Средообразующая функция. Живое вещество преобразует физико-химические параметры среды в условия, благоприятные для существования организмов. В этом проявляется еще одна главная функция живого вещества – средообразующая. Например, леса регулируют поверхностный сток, увеличивают влажность воздуха, обогащают атмосферу кислородом.
Можно сказать, что средообразующая функция – совместный результат всех рассмотренных выше функций живого вещества: энергетическая функция обеспечивает энергией все звенья биологического круговорота; деструктивная и концентрационная способствуют извлечению из природной среды и накоплению рассеянных, но жизненно важных для организмов элементов. Средообразующие функции живого вещества создали и поддерживают баланс вещества и энергии в биосфере, обеспечивая стабильность условий существования организмов, в том числе человека. Вместе с тем живое вещество способно восстанавливать условия обитания, нарушенные в результате природных катастроф или антропогенного воздействия.
В результате средообразующей функции в географической оболочке произошли следующие важнейшие события: был преобразован газовый состав первичной атмосферы; изменился химический состав вод первичного океана; образовалась толща осадочных пород в литосфере; на поверхности суши возник плодородный почвенный покров (также плодородны воды океана, рек и озер). Живое вещество выполняет следующие биогеохимические функции: газовые, концентрационные, окислительно-восстановительные, биохимические и биогеохимические, связанные с деятельностью человека.
6. Молекулярные основы эволюции (белки, нуклеиновые кислоты, первичный и вторичный генетический коды)
Белки, высокомолекулярные природные полимеры, построенные из остатков аминокислот, соединенных амидной (пептидной) связью – СО–NH– Каждый белок. характеризуется специфической аминокислотной последовательностью и индивидуальной пространственной структурой (конформацией). На долю белков приходится не менее 50% сухой массы орг. соединений животной клетки. Функционирование белков лежит в основе важнейших процессов жизнедеятельности организма. Обмен веществ (пищеварение, дыхание и др.), мышечное сокращение, нервная проводимость и жизнь клетки в целом неразрывно связаны с активностью ферментов. Основу костной и соединительной тканей, шерсти, роговых образований составляют структурные белки. Они же формируют остов клеточных органелл (митохондрий, мембран и др.). Расхождение хромосом при делении клетки, движение жгутиков, работа мышц животных и человека осуществляются по единому механизму при посредстве белков сократительной системы. Важную группу составляют регуляторные белки, контролирующие биосинтез белков и нуклеиновых кислот
По составу белки делят на простые, состоящие только из аминокислотных остатков, и сложные. Сложные могут включать ионы металла или пигмент. В соответствии с формой молекул белки подразделяют на глобулярные и фибриллярные. Молекулы первых свернуты в компактные глобулы сферической или эллипсоидной формы, молекулы вторых образуют длинные волокна (фибриллы) и высокоасимметричны. Большинство глобулярных белков, в отличие от фибриллярных, растворимы в воде.
Строение белковых молекул. Практически
все белки построены из 20![]() аминокислот,
принадлежащих, за исключением глицина, к L-ряду. Аминокислоты соединены между
собой пептидными связями, образованными карбоксильной и
аминокислот,
принадлежащих, за исключением глицина, к L-ряду. Аминокислоты соединены между
собой пептидными связями, образованными карбоксильной и![]() аминогруппами
соседних аминокислотных остатков (см. формулу I):
аминогруппами
соседних аминокислотных остатков (см. формулу I):
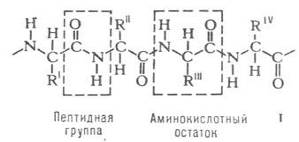 (формула I)
(формула I)
Белковая молекула может состоять из одной или нескольких цепей, содержащих от 50 до нескольких сотен (иногда – более тысячи) аминокислотных остатков. Молекулы, содержащие менее 50 остатков, часто относят к пептидам.
Различают
четыре уровня организации белковых молекул. Последовательность аминокислотных
остатков в полипептидной цепи наз. первичной структурой. Все белки различаются
по первичной структуре; потенциально возможное их число практически неограниченно.
Термин «вторичная структура» относится к типу укладки полипептидных цепей. Наиболее
часто встречающиеся типы – правая![]() спираль и
спираль и![]() структура. Под третичной структурой белков понимают
расположение его полипептидной цепи в пространстве. Термин «четвертичная
структура» относится к макромолекулам, в состав которых входит несколько
полипептидных цепей, не связанных между собой ковалентно. Такая структура
отражает способ объединения и расположения этих субъединиц в пространстве.
структура. Под третичной структурой белков понимают
расположение его полипептидной цепи в пространстве. Термин «четвертичная
структура» относится к макромолекулам, в состав которых входит несколько
полипептидных цепей, не связанных между собой ковалентно. Такая структура
отражает способ объединения и расположения этих субъединиц в пространстве.
Свойства. Физ.-хим. свойства белков определяются их высокомолекулярной природой, компактностью укладки полипептидных цепей и взаимным расположением остатков аминокислот
Синтез. Биосинтез белков происходит в результате трансляции в субклеточных частицах – рибосомах, представляющих собой сложный рибонуклеопротеидный комплекс. Информация о первичной структуре белка «хранится» в соответствующих генах – участках ДНК – в виде последовательности нуклеотидов. В процессе транскрипции эта информация с помощью фермента – ДНК – зависимой РНК – полимеразы – передается на матричную рибонуклеиновую кислоту, которая, соединяясь с рибосомой, служит матрицей для синтеза белка. Выходящие из рибосомы синтезированные полипептидные цепи, самопроизвольно сворачиваясь, принимают присущую данному белку конформацию, а также подвергаются модификации благодаря реакциям различных функциональных групп аминокислотных остатков и расщеплению пептидных связей.
Значение белков в питании. Белки – необходимая составная часть продуктов питания. В процессе пищеварения Б. подвергаются гидролизу до аминокислот, которые и всасываются в кровь. Пищевая ценность белков зависит от их аминокислотного состава, содержания в них, так называемых, незаменимых аминокислот, не синтезирующихся в организмах. В питательном отношении растительные белки менее ценны, чем животные; они беднее лизином, метионином и триптофаном, труднее перевариваются. Наряду с этим выводят новые сорта растений, содержащие гены, ответственные за синтез недостающих аминокислот. Перспективно использование для этого методов генетической инженерии.
Нуклеиновые кислоты (полинуклеотиды), биополимеры, осуществляющие хранение и передачу генетической информации во всех живых организмах, а также участвующие в биосинтезе белков.
Первичная структура нуклеиновых кислот (Н.к.) представляет собой последовательность остатков нуклеотидов. Последние в молекуле Н.к. образуют неразветвленные цепи. В зависимости от природы углеводного остатка в нуклеотиде Н.к. подразделяют соответственно на дезоксирйбонуклеиновые (ДНК) и рибонуклеиновые (РНК) кислоты. В молекуле ДНК гетероциклы, входящие в остаток нуклеотида, представлены двумя пуриновыми основаниями – адeнином (А) и гуанином (G), и двумя пиримидиновыми основаниями – тимином (Т) и цитозином (С); РНК вместо Т содержит урацил (U). Кроме того, в Н.к. в небольших количествах обнаруживаются модифицированные остатки нуклеозидов – минорные нуклеозиды.
Свойства ДНК и РНК различны. Так, РНК легко расщепляется щелочами до мононуклеотидов, в то время как в тех же условиях стабильны. Это структурное различие определяет и меньшую устойчивость к воздействию кислот N‑гликозидных связей (связь между гетероциклом и остатком рибозы) в ДНК по сравнению с РНК.
Дсзоксирибонуклепновые кислоты. Нуклеотидный состав ДНК подчиняется ряду правил (правила Чаргаффа), важнейшее среди которых – одинаковое содержание А и Т, G и С у любой клеточной ДНК. Нуклеотидный состав РНК подобным правилам не подчиняется.
Пространствю структура ДНК описывается как комплекс двух полинуклеотидных антипараллельных цепей (рис. 1), закрученных относительно общей оси. Комплементарное спаривание А с Т и G с С осуществляется посредством водородных связей.
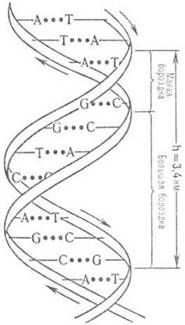
Рис. 1. Двойная спираль ДНК (стрелками показано направление полинуклеотидной цепи)
Установлено, что молекула ДНК в клетке представляет собой совокупность генов, регуляторных участков, районов, участвующих в организации генов в хромосомах.
Рибонуклеиновые кислоты. РНК, как правило, построены из одной полинуклеотидной цепи, характерный элемент вторичной структуры которой – «шпильки», перемежающиеся однотяжевыми участками (рис. 2). Шпилька – двутяжевая спиральная структура, образующаяся в результате комплементарного спаривания оснований (А с U и G с С). Шпильки и соединяющие их однотяжевые участки РНК укладываются в компактную третичную структуру. Для РНК вторичная структура имеет характерную форму, которую называют «клеверным листом». Известны редкие примеры целиком двухспиральных молекул РНК.
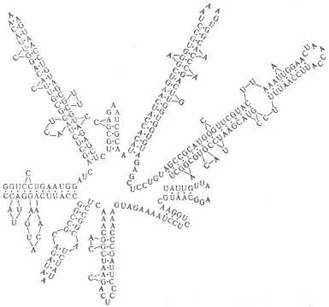
Рис. 2
Получение Н.к. В клетках Н.к. связаны с белками, образуя нуклеопротеиды. Выделение Н.к. сводится преимущественно к очистке их от белков. Для этого препараты, содержащие Н.к., обрабатывают ПАВ и экстрагируют белки фенолом. Последняя очистка и фракционирование Н.к. проводятся с помощью ультрацентрифугирования, различных видов жидкостной хроматографии и гель-электрофореза. Для получения индивидуальных Н.к. обычно используют различные варианты последнего метода.
Историческая справка. Н.к. открыты в 1869–72 Ф. Мишером в ядрах (отсюда назв.: лат. nucleus‑ядро) клеток гноя и в сперме лосося. В 1889 Р. Альтман выделил их в чистом виде (им же предложен термин «Нуклеиновые кислоты»). В 1944 О. Эйвери показал, что с помощью ДНК наследственные признаки могут быть переданы от одной клетки к другой и что ДНК, таким образом, является «веществом наследственности». Хим. строение Н.к. изучалось школами А. Косселя, П. Левина, Дж. Гулленда и А. Тодда и было окончательно установлено к нач. 50‑х гг. Макромол. Структура ДНК (двойная спираль) установлена в 1953 Дж. Уотсоном и Ф. Криком на основании данных рентгеноструктурного анализа, полученных Р. Франклин и М. Уилкинсом.
Генетический код, система «записи» наследственной информации в виде последовательности нуклеотидов в молекулах нуклеиновых кислот. Реализация генетического кода в клетке происходит в два этапа: 1) синтез молекулы матричной, или информационной, мРНК на соответствующем участке ДНК; при этом последовательность нуклеотидов ДНК «переписывается» в нуклеотидную последовательность мРНК; 2) синтез белка, при котором последовательность нуклеотидов РНК переводится в соответствующую последовательность аминокислот.
Впервые идея о существовании генетического кода сформулирована А. Дауном и Г. Гамовым в 1952–54, которые показали, что последовательность нуклеотидов, однозначно определяющая синтез той или иной аминокислоты, должна содержать не менее трех звеньев. Позднее было доказано, что такая последовательность состоит из трех нуклеотидов, названных кодоном, или триплетом. Т.к. молекулы нуклеиновых кислот, на которых происходит синтез мРНК или белка, состоят из остатков только четырех разных нуклеотидов, кодонов, отличающихся между собой.
Все синтезируемые в процессе трансляции белки построены из остатков 20 аминокислот (т. наз. кодируемых). Какой именно кодон ответствен за включение той или иной аминокислоты, можно определить по таблице, в которой буквы А, Г, У, Ц обозначают основания, входящие в нуклеотиды (соотв. аденин, гуанин, урацил, цитозин): в вертикальном ряду слева – в первый нуклеотид кодона, в горизонтальном ряду сверху – во второй, в вертикальном ряду справа – в третий. Трехбуквенные сочетания, напр. фен, сер, лей – сокращенные названия аминокислот. Прочерки в таблице означают, что три кодона – УАА, УАГ и УГА в нормальных условиях не кодируют какие-либо аминокислоты. Такие кодоны называются «бессмысленными», или нонсенс-кодонами. Они являются «сигналами» остановки синтеза полипептидной цепи.
Таблица генетического кода
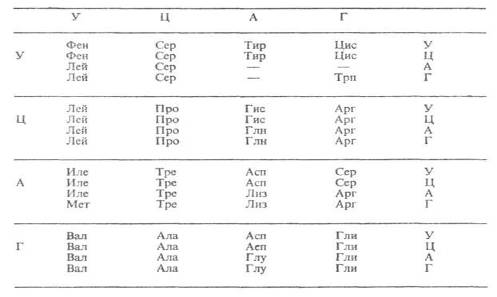
Г.к. называют вырожденным, поскольку 61 кодон кодирует всего 20 аминокислот. Поэтому почти каждой аминокислоте соответствует более чем один кодон. Вырожденность г.к. неравномерна: для аргинина, серина и лейцина она шестикратна, тогда как для мн. др. аминокислот (тирозина, гистидина, фенилаланина и др.) лишь двукратна. Кодоны-синонимы почти всегда отличаются друг от друга по последнему из трех нуклеотидов, тогда как первые два совпадают. Код аминокислоты определяется в основном первыми двумя «буквами». Вырожденность Г. к. имеет важное значение для повышения устойчивости генетической информации.
С механизмами трансляции связана еще одна особенность Г.к.: он неперекрывающийся. Кодоны транслируются всегда целиком; для кодирования невозможно использование элементов одного из них в сочетании с элементами соседнего. «Рамкой», ограничивающей транслируемый кодон и перемещающейся скачком сразу на три нуклеотида, служит антикодон тРНК. Наблюдается линейное соответствие между последовательностью кодирующих триплетов и расположением остатков аминокислот в синтезируемом полипептиде, т.е. код имеет линейный непрерывающийся порядок считывания.
Важнейшее свойство Г.к. – его однонаправленность. Кодоны информативны только в том случае, если они считываются в одном направлении – от первого нуклеотида к последующим.
Заключение
Химическая эволюция на Земле создала все предпосылки для появления живого из неживой природы. А Земля оказалась в таких специфических условиях, что эти предпосылки смогли реализоваться. Жизнь во всем ее многообразии возникла на Земле самопроизвольно из неживой материи, она сохранилась и функционирует уже миллиарды лет. Жизнь полностью зависит от сохранения соответствующих условий ее функционирования. А это во многом зависит от самого человека. Видимо, одним из проявлений природы и является появление человека как самосознающей себя материи. На определенном этапе он может оказывать ощутимое воздействие на среду собственного обитания, причем как позитивное, так и негативное.
Список используемой литературы
1) Концепции современного естествознания: Учебник для вузов / В.Н. Лавриненко, В.П. Ратников, Г.В. Баранов и др.; Под ред. проф. В.Н. Лавриненко, В.П. Ратникова. – 2-е изд., перераб. и доп. – М.: ЮНИТИ-ДАНА, 1999. – 303 с.
2) Концепции современного естествознания: Учебник. – Изд. 2-е, перераб. и доп. – М.: Альфа‑М, 2004. – 622 с. (в пер.)
3) Концепции современного естествознания: Учеб. пособие для вузов / А.А. Горелов. – М.: ООО «Издательство Астрель»: ООО «Издательство АСТ», 2003. – 380, [4] с. – (Высшая школа).
4) Пуассон А. Теории и символы алхимиков // Теории и символы алхимиков. М., 1995. С. 36.
5) http://www.xumuk.ru/encyklopedia/2/2974.html
6) http://www.xumuk.ru/encyklopedia/2/3846.html
7) http://www.xumuk.ru/encyklopedia/2665.html
8) http://www.xumuk.ru/encyklopedia/486.html
9) http://www.xumuk.ru/encyklopedia/970.html
[1] Пуассон А. Теории и символы алхимиков // Теории и символы алхимиков. М., 1995. С. 36.