Рефераты по авиации и космонавтике
Рефераты по административному праву
Рефераты по безопасности жизнедеятельности
Рефераты по арбитражному процессу
Рефераты по архитектуре
Рефераты по астрономии
Рефераты по банковскому делу
Рефераты по сексологии
Рефераты по информатике программированию
Рефераты по биологии
Рефераты по экономике
Рефераты по москвоведению
Рефераты по экологии
Краткое содержание произведений
Рефераты по физкультуре и спорту
Топики по английскому языку
Рефераты по математике
Рефераты по музыке
Остальные рефераты
Рефераты по биржевому делу
Рефераты по ботанике и сельскому хозяйству
Рефераты по бухгалтерскому учету и аудиту
Рефераты по валютным отношениям
Рефераты по ветеринарии
Рефераты для военной кафедры
Рефераты по географии
Рефераты по геодезии
Рефераты по геологии
Рефераты по геополитике
Рефераты по государству и праву
Рефераты по гражданскому праву и процессу
Рефераты по кредитованию
Рефераты по естествознанию
Рефераты по истории техники
Рефераты по журналистике
Рефераты по зоологии
Рефераты по инвестициям
Рефераты по информатике
Исторические личности
Рефераты по кибернетике
Рефераты по коммуникации и связи
Рефераты по косметологии
Рефераты по криминалистике
Рефераты по криминологии
Рефераты по науке и технике
Рефераты по кулинарии
Рефераты по культурологии
Статья: Incorporation of [2,3,4,5,6-2H5]Phenylalanine, [3,5-2H2]Tyrosine, and [2,4,5,6,7-2H5]Tryptophan into the Bacteriorhodopsin Molecule of Halobacterium halobium
Статья: Incorporation of [2,3,4,5,6-2H5]Phenylalanine, [3,5-2H2]Tyrosine, and [2,4,5,6,7-2H5]Tryptophan into the Bacteriorhodopsin Molecule of Halobacterium halobium
Applied Biochemistry and Microbiology, Vol. 35, ffo. /. 1999, pp. 29-17. Translated from Prikladnayti Biokhimiya i Mikrobialogiya, Vol. 35, No. 1,@ 1999, pp. 34-42. Original Russian Text Copyright © /999 hy Mosin, Skluclnev, Shvatz.
Incorporation of [2,3,4,5,6-2H5]Phenylalanine,
[3,5-2H2]Tyrosine, and [2,4,5,6,7-2H5]Tryptophan
into the Bacteriorhodopsin Molecule of Halobacterium halobium
O. V. Mosin*, D. A. Skladnev**, and V. I. Shvets*
* Lotnonosov Moscow State Academy of Fine Chemical Technology, Moscow, 117571 Russia
** State Center of Genetics and Selection of Industrial Microorganisms (GNU GENETICA), Moscow, 113515 Russia
Received September 25, 1997
Abstract—Incorporation of [2,3A5,6-2H5]phenylalanine, [3,5-2H2]tyrosine, and [2,4,5,6,7-2H5]tryptophan into the bacteriorhodopsin molecule followed by semipreparative isolation of bacteriorhodopsin resulted in a yield of 8-10 mg per g bacterial biomass. This method is based on the growth of the strain of halophilic bacteria Halobacterium halobium on a synthetic medium containing 2H-labeled aromatic ammo acids and fractionation of solubilized (in 0.5% sodium dodecyl sulfate) protein by methanol, including purification of carotenoids. lip-ids, and high-molecular-weight and low-molecular-weight compounds, as well as gel-permeation chromatog-raphy on Sephadex G-200. Incorporation of 2H-labeled amino acids was analyzed by electron impact mass spectrometry after hydrolysis of the protein in 4 N Ba(OH)2 and separation in the form of methyl esters of /V-DNS derivatives of amino aids by re versed-phase high-performance liquid chromatography.
The retinal-containing protein (a chromophore, pro-tonated aldimine of retinal containing Lys-216 e-amino group) bacteriorhodopsin (BR), functioning as an ATP-dependent translocase in cell membranes of halophilic bacteria Halobacterium halobium was initially described by Oesterhelt [1]. In spite of the fact that the structure and functions of this protein were studied in detail, it is still a focus of interest. This protein is used in practice as a biological photochromic material because of its high photosensitivity and resolution abilities [2]. Moreover, BR is attractive as a model object for studies of the functional activity and structural properties of membrane proteins hi the composition of artificially designed energy-transforming membranes.
The introduction of isotopic labels into molecules of membrane proteins is appropriate for studies of these proteins. Isotopic labels allow using the method of high-sensitivity electron impact (El) mass spectrometry for further analysis of isotopic incorporation [3, 4]. Thus, studies of BR labeled with the hydrogen isotope (deuterium) at residues of functionally important amino acids (phenylalanine, tyrosine, and tryptophan) involved in hydrophobic interaction of the protein polypeptide chain with the lipid bilayer of the cell membrane are important for practice [5, 6]. Raw 2H-labeled amino acids can be readily synthesized in preparative quantities by a reverse isotopic 1H-2H exchange in molecules of protonated amino acids, [2,3,4,5,6-2H5]phenylalanine (in 85% 2H2SC>4 at50°C), [3,5-2H2]tyrosine (in 6 N 2H2SO4 at slight boiling), and [2,4,5,6,7-2H5]tryptophan (in 75% [2H]trifluoroacetic acid at 25°C) [7]. However, in spite of the rapid development of chemical methods for obtaining 2H-labeled
aromatic amino acids, the Russian industry of individual 2H-labeled membrane proteins has not received wide acceptance.
This work was designed to obtain sernipreparative quantities of 2H-labeled BR for reconstruction of artificial membranes. Processes of incorporation of [2,3,4,5,6-2H5]phenylaIanine, [3,5-2H2]tyrosine, and [2,4,5,6,7-2H5]tryptophan into the molecule of bacteriorhodopsin followed with further semipreparative isolation were performed. The deuteration level was determined by means of El mass spectrometry performed after separation of the protein hydrolysate in the form of methyl esters of /V-DNS derivatives of amino aids by reverse-phase high-performance liquid chromatography (HPLC).
MATERIAL AND METHODS
Objects of studies. The carotenoid-contain ing strain of extreme halophilic bacteria Halobacterium halo-bium ET 1001 from the collection of cultures of microorganisms (Moscow State University) was used. The strain was maintained on solid peptone medium (2% agar) containing 4.3 M NaCl.
Preparation of growth media. DL-amino acids (Reanal, Hungary), adenosine monophosphate (AMP) and uridine monophosphate (UMP) (Sigma, USA), were used. 5-[Dimethylamino]naphthalene-l-sulfonyl chloride (DNS chloride; Sigma, USA) and diaz-omethane obtained from JV-nitroso-Af-methylurea (Merck, Germany) were applied for the synthesis of amino acid derivatives. [2,3,4,5,6-2H5]Phenylalanine (90 at. % 2H), [3,5-2H2]tyrosine (96 at. % 2H), and
[2,4,5,6,7-2H5]tryptophan (98 at. % 2H) (methods for obtaining are described in [8, 9]) were supplied by A.B. Pshenichnikova (Candidate of Chemical Sciences, Lomonosov Moscow State Academy of Fine Chemical Technology).
2H-Labeled BR. 2H-Labeled BR was obtained on a synthetic medium, in which protonated ammo acids (phenylalanine, tyrosine, and tryptophan) were replaced by their deuterium-containing analogues ([2,3,4,5,6-2H5]phenylalanine, [3,5-2H2]tyrosine, and [2,4,5,6,7-2HJtryptophan). The medium contained 0.43 g/1 DL-alanine, 0.4 g/1 L-arginine,0.45 g/1 DL-aspartic acid, 0.05 g/1 L-cysteine, 1.3 g/1 L-glutamic acid, 0.06 g/1 L-glycine, 0.3 g/1 DL-histidine, 0.44 g/1 DL-isoleucine, 0.8 g/1 L-leucine, 0.85 g/1 L-lysine, 0.37 g/1 DL-methionine, 0.26 2/1 DL-phenylalanine, 0.05 g/1 L-proline, 0.61 g/1 DL-serine, 0.5 g/1 DL-thre-onine, 0.2 g/1 L-tyrosine, 0.5 g/1 DL-tryptophan, 1.0 g/1 DL-valine, nucleotides (0.1 g/1 AMP and 0.1 g/1 UMP), salts (250 g/I Nad, 20 g/1 MgSOa x 7H2O, 2 g/1 KC1, 0.5 g/1 NH4C1, 0.1 g/1 KNO3, 0.05 g/1 KH2PO4, 0.05 g/1 KoHPO4, 0.5 g/1 sodium citrate, 3 x 10 -4 g/1 MnSO4 x 2H2O, 0.065 g/1 CaCl2 - 6H2O, 4 x 10 -5 g/l ZnSO4 x 7H2O, 5 x 10 -5FeSO4 - 7H2O, and 5 x 10 -5 g/1 CuSO4 x 5H2O), 1 g/1 glycerin, and growth factors (1 x 10 -4 g/1 biotin, 1.5 x l0 -4 g/1 folic acid, and 2 x 10 -5 g/1 vitamin B!2).
Cultivation of bacteria. The growth medium was autoclaved for 30 min at 0.5 atm (pH was brought to 6.5-6.7 using 0.5 N KOH). The inoculum was grown in 750-ml Erlenmeyer's flasks (the medium volume was 100 ml) on a 380-S orbital shaker (Biorad, Hungary) at 35-37°C under conditions of intensive aeration and illumination (three LDS-40 lamps of 1.5 Ix each). After 24 h, the inoculum (5-10%) was transferred to the synthetic medium and grown for five to six days (similarly to obtaining of the inoculum). All further manipulations for BR isolation were performed with the use of a dimming lamp equipped with an ORZh-1 orange light filter.
Isolation of the fraction of purple membranes (PM). The biomass (1 g) was washed with distilled water and precipitated on a T-24 centrifuge (Carl Zeiss, Germany) at 1500 g for 20 min. The precipitate was suspended in 100 ml of distilled water and kept at 4°C. After 24 h, the reaction mixture was centrifuged at 1500 g for 15 min. The precipitate was resuspended in 20 ml of distilled water, disintegrated by sonication (2 kHz, three times per 5 min) on a water bath containing ice (0°C), and centrifuged at 1500 g for 20 min. After washing with distilled water, the cellular homogenate was resuspended in 10 ml of buffer containing 125 mM NaCl, 20 mM MgCl2, and 4 mM Tris-HCl (pH 8.0). RNase (5 u,g, two-three units of activity) was added. The mixture was incubated at 37°C. The same buffer (10 ml) was added 2 h later. The mixture obtained was kept at 4°C for 14-16 h. The water fraction was removed by centrifugation at 1500 g for 20 min. The precipitate of
PMs was treated (five times) with 7 ml of 50% ethanol at -5°C. The solvent was removed by centrifugation at 1200 g and cooling for 15 min. The protein concentration was measured on a DU-6 spectrophotometer (Beckman, USA) calculating the D280/D56S ratio [10]. Regeneration of PMs was conducted as described in [11].
Isolation of BR. The fraction of PMs (1 mg/ml) was solubilized in 1 ml of 0.05% sodium dodecyl sulfate (SDS), kept at 37°C for 7-9 h, and centrifuged at 1200 g for 15 min. The precipitate was removed. Methanol (100 (ll) was added drop wise (three times) to the supernatant at 0°C. The mixture was kept at -5°C for 14-15 h and then centrifuged at 1200 g and cooling for 15 min. Fractionation was performed three times with decreasing the concentration of SDS to 0.2% and 0.1%. Crystalline protein (8-10 mg) was washed with cold distilled water and centrifuged at 1200 g for 15 min.
Purification of BR. This procedure was performed by gel-permeation chromatography on a calibrated column (150 x 10 mm). Sephadex G-200 (Pharmacia, USA) served as the stationary phase (bed volume: 30-40 ml per g). The samples were taken manually. The column was balanced with the buffer solution containing 0.1% SDS and 2.5 mM EDTA. The protein sample was dissolved in 100 p.1 of the buffer solution and eluted with 0.09 M Tris-borate buffer (pH 8.5, / = 0.075) and 0.5 M NaCl at a flow rate of 10 ml/cm2 per h. Combined protein fractions were subjected to lyo-philization.
Electrophoresis of the protein. The procedure was performed in 12.5% polyacrylamide gel (PAAG) containing 0.1% SDS. The samples were prepared for elec-trophoresis by standard procedures (LKB protocol, Sweden). Electrophoretic gel stained with Coomassie blue R-250 was scanned on a CDS-200 laser densitom-eter (Beckman, USA) for quantitative analysis of the protein level.
Hydrolysis of BR. The protein (4 mg) was placed into glass ampoules (10 x 50 mm in size), and 4 N Ba(OH)2 (5 ml) was added. The mixture was kept at 110°C for 24 h. The reaction mixture was suspended in 5 ml of hot distilled water and neutralized with 2 N H2SO4 to pH 7.0. The sediment of BaSO4 was removed by centrifugation at 200 g for 10 min, and the supernatant was evaporated in a rotor evaporator at 40°C.
Synthesis of N-DNS derivatives of amino acids. DNS chloride (25.6 mg) in 2 ml of acetone was added gradually to 4 mg of dry hydrolysate of BR in 1 ml of 2 M NaHCO3 (pH 9-10) under conditions of constant mixing. The reaction mixture was kept at 40°C and mixing for 1 h, acidified with 2 N HCI to pH 3, and extracted (three times) with 5 ml of ethyl acetate. The combined extract was washed with distilled water to pH 7.0 and dried with anhydrous Na2SO4. The solvent was removed at 10 mmHg.
Methyl esters of N-DNS derivatives of amino acids. Wet N-nitroso-.N'-methylurea (3 g) was added to 20 ml of 40% KOH in 40 ml of diethyl ether and then mixed
on a water bath with ice for 15-20 min for obtaining diazomethane. After the completion of gas release, the ether layer was separated, washed with distilled water to pH 7.0, dried with anhydrous Na2SO4, and used for the treatment of /V-DNS derivatives of amino acids.
Separation of the mixture of methyl esters ofN-DNS derivatives of amino acids. This was performed by the method of reverse-phase high-performance liquid chro-matography on a Knauer liquid chromatograph (Germany) equipped with a Knauer pump, 2563 UV detector, and C-R 3A integrator (Shimadzy, Japan). The column of 250 x 10 mm in size was used. Separon C18 (Kova, Czech) served as the stationary reverse phase. The diameter of granules was 12 urn. The injection volume was 10 mkl. The following systems of solvents were used: (A) acetonitrile and trifluoroacetic acid (at a volume ratio of 100 : 0.1-0.5) and (B) acetonitrile. Gradient elution processes were performed at a rate of 1.5 ml/min for 5 min (from 0% to 20% B), 30 min (from 20% to 100% B), 5 min (100% B), 2 min (from 100% to 0% B), and 10 min (0% B).
Mass spectra. Mass spectra of methyl esters of N-DNS derivatives of amino acids were obtained by the method of electron impact on an MB-80 A instrument (Hitachi, Japan) at the energy of ionizing electrons of 70 eV, accelerating potential of 8 kV, and a temperature of the cathode source of 180-200°C. Scanning of the samples analyzed was performed at a resolution of 7500 conditional units and a 10% image definition.
RESULTS AND DISCUSSION
Incorporation of [2,3,4,5,6-2H5]phenylalanine, [3,5-2H2]tyrosine, and [2,4,5,6,7-2H5]tryptophan into the molecule of BR. The method of incorporation of 2H-labeled amino acids into the molecule of BR was selected because of the fact that this work was designed to reveal the possibility for obtaining 2H-labeled preparations of the membrane protein (in semipreparative amounts) for the reconstruction of artificial membranes. [2,3,4,5,6-2H5]PhenyIalanine, [3,5-2H2]ryrosine, and [2,4,5,6,7-2H5;]tryptophan play important roles in hydrophobic interaction of the BR molecule with the lipid bilayer of the cell membrane. They are stable to the 'H-2H exchange in water medium under growth conditions. Moreover, high-sensitivity El mass spec-trometry can be used for the analysis of their incorporation, which was performed microbio logically by growing the strain of halophilic bacteria Halobacte-rium halobium on a synthetic medium containing 2H-labeled aromatic amino acids. Thus, these compounds were selected as sources of deuterium. Under the optimum growth conditions (exponential growth on a synthetic medium with 4.3 M NaCl at 35-37°C and illumination), the cells synthesized a purple pigment whose spectral characteristics were identical to those of native BR. Figure 1 shows the dynamics of (2) bacterial growth on the medium containing -H-labeled aromatic amino acids in relation to (1) growth under control con-
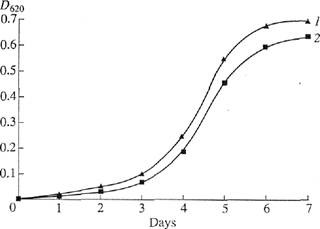
Fig. 1. The dynamics of Che growth of Che strain//, halobium under various experimental conditions: (/) protonated synthetic medium and (2) synthetic medium with [2,3,4,5,6-2H5]phenylalanine, [3,5-2H2Jtyrosine, and [2,4,5,6,7-2H5]tryptophan.
ditions. The growth of this strain on the medium containing 2H-Iabeled aromatic amino acids was only slightly inhibited. This is important for producing the raw 2H-labeled biomass for further isolation of BR.
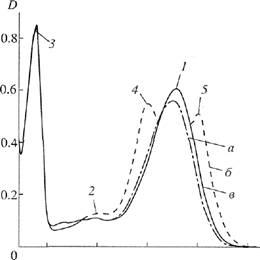
The main stages of isolating 2H-labeled BR (Fig, 2) were the following: production of 1 g of 2H-labeled bio-mass; isolation of the fraction of PMs; removal of low-molecular-weight and high-molecular-weight admixtures, cellular RNA, carotenoids, and lipids; fraction-ation of solubilized (in 0.05% SDS) protein by metha-nol; and purification on Sephadex G-200. Low-molecular-weight admixtures and the intracellular contents were eliminated by osmotic shock induced by distilled water (after removing 4,3 M NaCl) followed by destruction of cell membranes by ultrasound. The cellular homogenate was then treated with RNase I (two-three units of activity) to induce the maximum destruction of cellular RNA. The PM fraction obtained contained the complex of the desired protein with Hpids and polysaccharides, as well as admixtures of fixed carotenoids and foreign proteins. Therefore, it was necessary to use special methods of protein fracdonation, which would not damage the native structure of the protein native structure or cause its dissociation. This made the isolation of pure individual BR performed by the use of special fine methods for removing carotenoids and lipids, purification, and column chromatography more difficult. Decarotenoidation was conducted by a repeated treatment of PMs with 50% ethanol at -5°C. Although it was a routine procedure, this stage was necessary (despite of considerable chromoprotein losses). The treatment was repeated no less than five times to obtain the absorption band of the PM suspension freed of carotenoids. Figure 3 shows (curves b, c) these bands at various stages of treatment in relation to (curve a) the band of
Growth of Halobacterium halobium on synthetic medium containing [2,3,4,5,6-2H5]phenyIalanine, [3,5-2H2]tyrosine and [2,4,5,6,7-2H5]tryptophan
|
|
| Disintegration by ultrasound |
|
Water-soluble products of cellular content, inorganic salts, and other low-molecular-weight compounds |
 Distilled
H2O
Distilled
H2O
RNase I,
125 mM NaCl, 20 mM MgCl,
4 mM Tris-HCl
Distilled H2O
Isolation of the biomass
Raw
biomass
________ t
Osmotic shock
Culture liquid
4.3 M NaCl, and other
inorganic salts
and metabolites
50% ethanol
1.0.5%SDS-Na 2. Methanol
-5°C
-5°C
PM fraction
Decarotenoidation
±
Delipidation + BR precipitation
— Extract of carotenoids
_._ Residuals of cellular walls, lipids, and other high-molecular-weight compounds
Crystalline
BR
t
Gel-permeation chromatography on Sephadex G-200
4NBa(OH)7 UO°C,24h
1.
DNS
chloride, 2 M
NaHCO3, and ethyl acetate
2. jV-Nitroso-N- methyl-_
urea, 40% KOH
diethyl ester, and diazomethane
Purified BR ±
![]()
Mixture of free amino acids I
Modification into methyl esters
of /V-DNS derivatives of amino acids
Reverse-phase HPLC
BaSO4 after neutralization with 2 M 2 M H2SO4
Individual methyl esters of/V-DNS[2,3,4,5,6-2H5]phenylalanine
N-DNS-[3,5-2H2]tyrosine, and N-DNS [2,4,5,6,7-2H5]tryptophan
El mass spectrometry
Fig. 2. Experimentally designed method for isolating H-labeled BR.
native BR. In this case, an 80-85% efficiency of removing carotenoids was reached. The formation of the retinal-protein complex induced a bathochromatic shift in the absorption band of PMs (Fig. 3). The major band recorded at the maximum absorption of 568 nm and induced by the light isomerization of chromophore at
bonds positioned at C13=C14 or multiples of this number was determined by the presence of trans-retinal residue of retinal (BR568). The additional low-intensity band recorded at 412 nm characterized the presence of a minor admixture of the M412 spectral form (produced in light) containing the deprotonated aldirnine bond
 between the residue
of trans-retinal and the protein. The band recorded at
280 nm depended on the absorption of aromatic amino acids of the polypeptide chain of this
protein (the D2%0/D56% ratio was 1.5 :
1 for pure BR).
between the residue
of trans-retinal and the protein. The band recorded at
280 nm depended on the absorption of aromatic amino acids of the polypeptide chain of this
protein (the D2%0/D56% ratio was 1.5 :
1 for pure BR).
Fractionation and careful chromatographic purification of the protein were the next necessary stages. BR is a transmembrane protein with a molecular weight of 26.7 kDa that penetrates the lipid bilayer in the form of seven a-helixes. Therefore, the use of ammonium sul-fate and another traditional salt-eliminating agents is not appropriate. The protein must be transformed into the soluble form by solubilization in 0.5% SDS. The use of this ionic detergent was dictated by the necessity of the most complete solubilization of the protein achieved by combining delipidation and precipitation. In this case, BR solubilized in a low-concentration solution of SDS retained its helical cc-conformation [12]. Therefore, it was not necessary to use organic solvents such as acetone, methanol, and chloroform for removing lipids. Delipidation and precipitation of the protein were combined into the same stage. This noticeably simplified fracdonation. The advantage of this method was that the desired protein (in the complex with molecules of lipids and detergent) was in the supernatant. Another high-molecular-weight admixtures were in the nonreacted precipitate, which was removed by centrifugation. Fractionation of solubilized (in 0.5% SDS) protein and its further isolation in the crystalline form were conducted using a gradual low-temperature (-5°C) precipitation by methanol (three stages). The second and the third stages were performed by decreasing the detergent concentration 2.5 and 5 times, respectively. The final stage of BR purification involved the separation of the protein from low-molecular-weight admixtures by gel-permeation chro-matography. The fractions containing BR were passed two times through a column with dextran Sephadex G-200 balanced with 0.09 M Tris-borate buffer (pH 8.35) containing 0.1% SDS and 2.5 mM EDTA. The method designed for fractionation of the protein made it possible to obtain 8-10 mg of pure preparation of 2H-labeled BR from 1 g of bacterial biomass. The homogeneity of BR complied with the requirements on reconstruction of membranes and was confirmed by electrophoresis in 12.5% PAAG with 0.1% SDS, regeneration of apomembranes with trans-retinal, and reverse-phase HPLC of methyl esters of N-DNS derivatives of amino aids. Low yield of BR was no barrier to further studies of isotopic incorporation. However, it must be emphasized that considerable amounts of the raw biomass must be produced in order to provide high yield of the protein.
Hydrolysis of BR. Conditions of hydrolysis of deuterium-containing protein were determined by the necessity of preventing the isotopic ('H-2H) hydrogen-deuterium exchange in molecules of aromatic amino acids, as well as retaining tryptophan in the protein hydrolysate. Two alternative variants (acid and alkaline hydrolysis) were considered. Acid hydrolysis of the
| 300 |
400 500 600 700
nm
Fig. 3. Absorption bands (in 50% ethanol) at various stages of treatment: (a) native BR, (b) PMs after intermediate treatment, and (c) P.Ms purified of foreign admixtures. The band (/) corresponds to the spectral form of BR568. The band (2) corresponds to the admixture of the M^ spectral form. The band (J) characterizes the absorption of aromatic amino acids. The bands (4) and (5) correspond to foreign caro-tenoids. Native BR was used as control.
protein performed under standard conditions (6 N HC1 or 8 N H2SO4, 110°C, 24 h) is known to induce complete degradation of tryptophan and partial degradation of serine, threonine, and several other amino acids in the protein [13]. These amino acids do not play an important role in this study. The modification of this method involving the addition of phenol [14], thiogly-colic acid [15], and p-mercaptoethanol [16] into the reaction medium allowed retaining tryptophan (to 80-85%). 7-ToIuenesulfonic acid with 0.2% 3-(2-aminoet-hyl)-indole, as well as 3 M 2-mercaptoethanesulfonic acid [18], are the potent agents for retaining tryptophan (to 93% [17]). However, these methods are not suitable for working the problem, because they have a noticeable weakness. Processes of the isotopic exchange (of a high rate) of aromatic protons (deuterons) in molecules of tryptophan, tyrosine, and histidine [19], as well as the exchange of protons at C3 atom of aspartic acid and C4 atom of glutamic acid [20], proceed under conditions of acid hydrolysis. Thus, the data on incorporation of deuterium into the protein can not be derived from the hydrolysis performed even in deuterium-containing reagents (2HC1,2H2SO4, and 2H2O).
Reactions of the isotopic hydrogen exchange are nearly undetected (except for the proton (deuteron) at C2 atom of histidine), and tryptophan is not degraded under conditions of alkaline hydrolysis (4 N Ba(OH)2 or NaOH, 110°C, 24 h). Thus, this method of hydroly: sis was used in our study. Simplification of the procedure for isolating the mixture of free amino acids (due
|
|
| 527 |
| 200 |
| 100 |
| 300 |
| 400 |
| 500 |
| 600 |
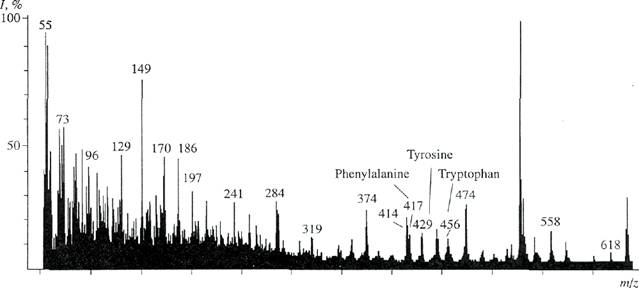
Fig. 4. El mass spectrum of the mixture of methyl esters of /V-DNS derivatives of amino acids of the BR hydrolysate. Cultivation was performed on synthetic medium containing [2,3,4,5,6- Hslphenylalanine, [3,5- H2]tyrosine, and [2,4,5,6,7-2H5]tryptophan. Images of molecular ions of arnino acids correspond to their derivatives (here and on Fig. 5). Ordinate: relative intensity of the peak /)-
to neutralization with H2SO4) was the cause of selection of 4 N Ba(OH)2 as a hydrolyzing agent. Possible racemization of amino acids during alkaline hydrolysis did not affect the results of further mass-spectrometry assay showing the deuteration level of molecules of amino acids.
Study of incorporation of [2,3,4,5,6-2H5]phenylala-nine, [3,5-2H2]tyrosine, and [2,4,5,6,7-2H5]tryptophan into the molecule ofBR. El mass spectrometry following the modification of the mixture of free amino acids of the protein hydrolysate into methyl esters of N-DNS derivatives of amino acids was used for studies of incorporation of 2H-labeled aromatic amino acids. Total El mass spectrum of the mixture of methyl esters of N-DNS derivatives of 2H-labeled amino acids was recorded to obtain reproducible data on the incorporation of 2H-labeled aromatic amino acids. The deuteration level of molecules was determined by calculating the difference between the values of heavy peaks of molecular ions [M]+ enriched with deuterium of derivatives of aromatic amino acids and their light unlabeled analogues. Methyl esters of N-DNS derivatives of aromatic amino acids were separated by reverse-phase HPLC, and El mass spectra of individual-amino acids were obtained. The El mass spectrum of the mixture of methyl esters of N-DNS derivatives of amino acids (scanning at m/z 50-640, the base peak of m/z 527, 100%) was of the continuous type (Fig. 4). The peaks (in the range from 50 to 400 on the scale of mass numbers) were represented by fragments of metastable ions, low-molecular-weight admixtures, and products of chemical modification of amino acids. 2H-labeled aromatic amino acids with mass numbers in the range
from 414 to 456 on the scale of mass numbers were the mixtures of molecules containing various numbers of deuterium atoms. Therefore, their molecular ions [M]+ were polymorphously split (depending on the number of hydrogen atoms in the molecule) into individual clusters displaying static sets of m/z values. Taking into account the effect of isotopic polymorphism, the deuteration level was determined from the most commonly encountered peak of the molecular ion [M]+ (which value was mathematically averaged by mass spectrometer) in each cluster (Fig. 4). Phenylalanyne had a peak of a molecular ion that corresponded to [M]+ and was 13% at m/z 417 (instead of [M]+ at m/z 412 for unlabeled phenylalanine; peaks of unlabeled amino acids are not represented here). Tyrosine had the peak of molecular ion that corresponded to [M]+ and was 15% at m/z 429 (instead of [M]+ at m/z 428). Tryptophan had a peak of a molecular ion that corresponded to [M]+ and was 11 % at m/z 456 (instead of [M]+ at m/z 451). Levels of deuteration corresponding to the increase in molecular weights were one (for tyrosine) and five (for phenylalanine and tryptophan) atoms of deuterium. These results showing deuteration levels of phenylalanine, tyrosine, and tryptophan are in agreement with data on the deuteration levels of initial amino acids. This indicates a sufficiently high potency of incorporation of 2H-labeled aromatic amino acids into the protein molecule. Thus, incorporation of 2H-labeled amino acids into the BR molecule was of a specific type. Deuterium was detected in all residues of aromatic amino acids. However, it should be stressed that there were [M]+ peaks of protonated and semideuterated analogues of phenylalanine with [M]+ at m/z 414 (20%), 415 (18%), and 416
|
|
| (a) |
|
170. 234. A 353 B81 |
| 100 |
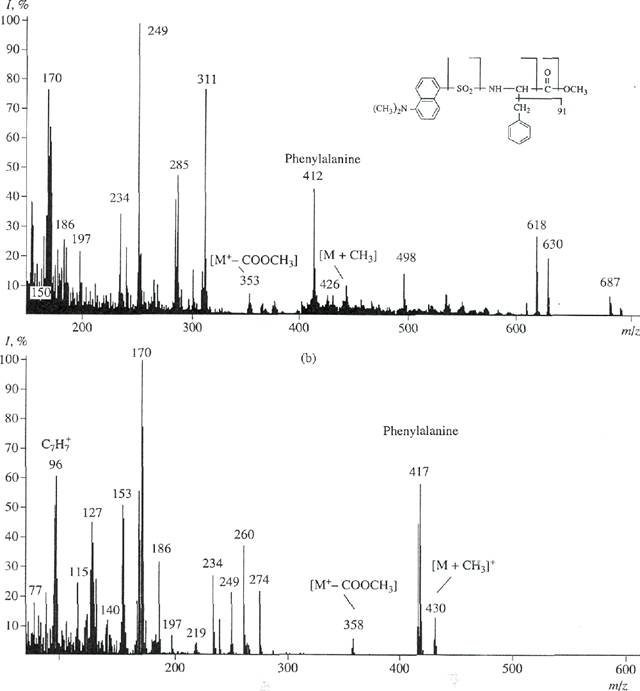
Fig, 5. El mass spectrum of the mixture of methyl esters of N-DNS phenylalanine under various experimental conditions: (a) unla-beled methyl ester of N-DNS phenylalanine and (b) methyl ester of /V-DNS [2,3,4,5,6-2H5] phenylalanine isolated by reverse-phase HPLC.
(11%); tyrosine with [M]+ at m/z428 (12%); and tryp-tophan with [M]+ at m/z 455 and 457 (9%) displaying various contributions to the deuteration level of molecules. This suggests that small part of minor pathways of their biosynthesis de novo leading to the dilution of a deuterium label was retained. The presence of these peaks probably depended on conditions of biosynthetic
incorporation of 2H-labeled aromatic amino acids into the protein molecule.
The analysis of scan El mass spectrum showed that peaks of molecular ions [M]+ of methyl esters of N-DNS derivatives of aromatic amino acids had low intensities and were polymorphously split. Therefore,
their molecular enrichment ranges were considerably
widened. Moreover, mass spectra of the mixture components were additive. Therefore, these mixtures can be analyzed only in the case of the presence of spectra of various components recorded under the same conditions. These calculations involve solution of the system of n equations in n unknowns for the mixture containing n components. For the components, whose concentrations are more than 10 mol %, the validity and repro-ducibility of the analysis results can be ±0.5 mol % at a confidence probability of 90%. Therefore, chromato-graphical isolation of individual derivatives of 2H-labeled amino acids from the protein hydrolysate is necessary for a obtaining a reproducible result. Reverse-phase HPLC on octadecylsilane silica gel, Separon C18 (whose potency was confirmed by separation of methyl esters of //-DNS derivatives of 2H-labeled amino acids of another microbial objects, e.g., methylotrophic bacteria and microalgae [21]), was used. This method was adapted to conditions of chro-rnatographical separation of a mixture of methyl esters of DNS derivatives of amino acids of the BR hydrolysate. Optimization of eluant ratios, the gradient type, and the rate of elution from the column were performed. The maximum separation was observed after gradient elution with a mixture of solvents containing acetonitrile and trifluoroacetic acid (at a volume ratio of 100 : 0.1-0.5). In this case, tryptophan and a hardly degraded pare of phenylalanine/tyrosine were successfully separated. Degrees of chromatographical purities of isolated methyl esters of N-DNS [2,3,4,5,6-2H5]phe-nylalanine, N-DNS [3,5-2H2]tyrosine, and N-DNS [2,4,5,6,7-2H5]tryptophan were 97%, 96%, and 98%, respectively. The yield was 97-85%. Figure 5b confirms the result obtained. This figure shows the El mass spectrum of methyl ester of N-DNS [2,3,4,5,6-2H5]phe-nylalanine isolated by reverse-phase HPLC (scanning at m/z 70-600; the base peak at m/z 170; 100%). The mass spectrum is represented in relation to unlabeled methyl ester of//-DNS phenylalanine (scanning at m/z 150-700; the base peak at m/z 250; 100%) (Fig. 5a). The peak of a heavy molecular ion of methyl ester of N-DNS phenylalanine ([M]+, 59% at m/z 417; instead of [M]+, 44% at m/z 412 for unlabeled derivative of phenylalanine) and the additional peak of the benzyl fragment of phenylalanine, C7H7 (61% at mlz 96; instead of 55% at mlz 91 for control; data not shown), confirm the presence of deuterium in phenylalanine. The peaks of secondary fragments of various intensities with m/z 249, 234, and 170 correspond to products of secondary degradation of the dansyl residue to N-dimethylaminon-aphthalene. The low-intensity peak of [M+-COOCH3] (7%) at m/z 358 (m/z 353, 10%, control) represents the detachment of the carboxymethyl group from methyl ester of N-DNS phenylalanine. The peak of [M + CH3]+ (15%) at m/z 430 (m/z 426, 8%, control) represents the additional methylation at a-amino group of phenylalanine. The difference between molecular weights of
light and heavy peaks of [M]+of methyl ester of N-DNS phenylalanine is five units. This is in agreement with the earlier obtained result and the data on the level of deutera-tion of initial [2,3,4,5,6-2H5]phenylalanine added into the growth medium.
Thus, these data indicate a high efficiency of incorporation of 2H-labeled aromatic amino acids into the BR molecule. Completely deuterated protein preparations for reconstruction (into 2H2O) of functionally active systems of membrane proteins with purified 2H-labeled lipids and other deuterated biologically active compounds are proposed to be obtained using the method elaborated. In the future, these studies will provide the means for solving the problem of functioning of 2H-Iabeled BR in the composition of artificially constructed membranes under conditions of deuterium-saturated medium.
ACKNOWLEDGMENTS
This work was supported by grant no. 1B-22-866 ("High chemical technologies"). We are grateful to Dr. B.M. Polanuer (GNU GENETICA) for careful attention and helpful remarks in discussions of the results.
REFERENCES
1.
Oesterhelt,
D. and Stoeckenius, W., Nature (London),
1971, vol. 233, no 89, pp. 149-160.
2.
Spudich, J.L., Ann. Rev.
Biophys. Chem, 1988, vol. 17,
no. 12, pp. 193-215.
3.
Karnaukhova,
E.N., Niessen, W.M.A., andTjaden, U.R.,
Anal
Biochem., 1989,
vol. 181, no. 3, pp. 271-275.
4.
Mosin,
O.V., Skladnev, D.A., Egorova, T.A., and
Shvets, V.I., Bioorg.
Khim., 1996, vol. 22, nos. 10-11,
pp. 856-869.
5.
Hardy,
J.R, Knight, A.E.W., Ghiggino, K.R, Smith, T.A.,
and Rogers,
P.J., Photochem. Photobiol, 1984, vol. 39,
no. 1, pp. 81-88.
6.
Rosenbach,
V., Goldberg, R., Gilon, C., and Ottolenghi, M.,
Photochem.
Photobiol, 1982,
vol. 36, no. 6, pp. 197-
201.
7.
Mosin,
O.V., Skladnev, D.A., Egorova, T.A., and
Shvets, V.I., Biotechnologiya, 1996,
no. 10, pp. 24-40.
8. Griffiths, D.V., Feeney, J., Roberts, G.C., and Burgen, A.S.,
Biochim. Biophys. Acta, 1976, vol. 446, no. 4, pp. 479-585.
9. Matthews, H.R.,
Matthews, K.S., and Opella, S.J., Bio
chim. Biophys. Acta, 1977, vol. 497, no. 23, pp. 1-13.
10. Oesterhelt,
D. and Hess, B., Eur. J. Biochem., 1973,
vol.
37, no. 1, pp. 316-326.
11.
Tokunada,
F. and Ebrey, T, Biochemistry, 1978, vol. 17,
no. 10, pp.
1915-1922.
12. Pervushin, K.V. and
Arsen'ev, A.S., Bioorg. Khim.,
1995, vol. 21, no. 10, pp.
83-111.
13. Zvonkova, E.N., Zotchik,
N.V., Filippovich, E.I., Mitro-
fanova, T.K.,
Myagkova, G.I., and Serebrennikova, G.A.,
Khimiya biologicheski aktivnykh
prirodnykh soedinenii
(Chemistry of Biologically Active Natural Compounds), Moscow: Khirniya, 1970, pp. 65-68.
14.
Muromoto,
K., Sunahara, S., and Kamiya, H., Agric.
Biol.
Chem., 1987,
vol. 51, no. 6, pp. 1607-1616.
15.
Matsubara,
H. and Sasaki, R.M., Biochim. Biophys. Res.
Com., 1969, vol. 35, no. 10, pp. 175-177.
16.
Ng,
L.T., Pascaud, A., and Pascaud, M., Anal. Biochem.,
1987, vol. 167, no. 2, pp.
47-52.
17.
Liu,
T.Y. and Chang, Y.H..J.Biol. Chem., 1971, vol. 246,
no. 2, pp.
2842-2848.
18.
Simpson,
R.J., Neuberger, MR., and Liu, T.Y., J. Biol.
Chem., 1976, vol. 251, no. 3, pp. 1936-1938.
19.
Pshenichnikova,
A.B., Karnaukhova, E.N., Zvonko
va, E.N., and
Shvets, V.I., Bioorgan. khimiya, 1995,
vol. 21, no.
3, pp. 163-178.
20.
Cohen,
J.S. and Putter, I., Biochim. Biophys. Acta, 1970,
vol. 222, no.
1, pp. 515-520.
2E. Egorova, T.A., Mosin, O.V., Eremin, S.V., Karnaukhova, E.N., Zvonkova, E.N., and Shvets, V.I., Bio-technologiya, 1993, no. 8, pp. 21-25.